The Office of Surveillance and Epidemiology in the Center for Drug Evaluation and Research (CDER) leads the Sentinel System. Sentinel was created to meet the mandate described in Section 905 of the Food and Drug Administration Amendments Act 2007 (FDAAA) to create an active postmarket drug safety surveillance system. CDER uses Sentinel to proactively assess the safety of FDA approved drugs under real-world conditions.
The Active Risk Identification and Analysis (ARIA) system is the largest and most developed component of Sentinel. ARIA uses state-of-the-art analysis tools and a distributed database of standardized claims and claims linked with electronic health records (EHR) data to monitor the safety of medications. The data undergo continuous quality checks and refreshes. With ARIA, safety analyses are conducted more efficiently compared to studies with fully customized analytics—often in a matter of months, rather than several years.
Providing evidence that alleviates a drug safety concern
To learn more about the activities and accomplishments of the Sentinel System from 2022 to 2024, please see the following assessment report. This report fulfills the Prescription Drug User Fee Act (PDUFA) VII commitment to “analyze, and report on the use of Sentinel for regulatory purposes.”
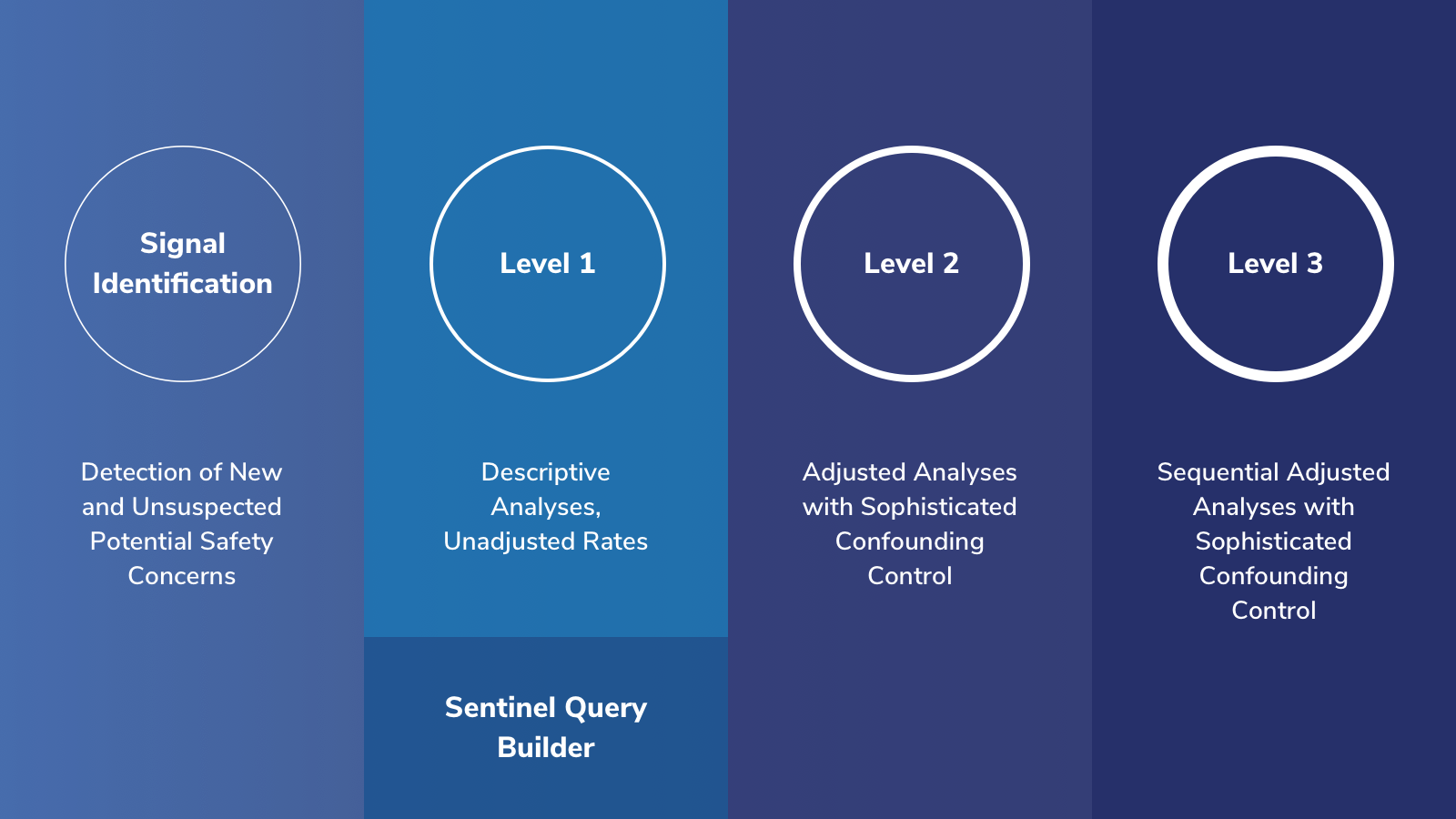
Signal Identification: analyses used to detect new and unsuspected potential safety concerns.
Level 1: analyses used to describe and characterize patterns of medication use or rates of health outcomes of interest. The Sentinel Query Builder application represents a subset of Level 1 functionality. Query Builder analyses are typically run on Merative™ MarketScan® Research Databases but some are also run on Sentinel's distributed database.
Levels 2 and 3: analyses used to study whether a potential adverse health outcome is related to the use of drug and estimates the size of that risk.
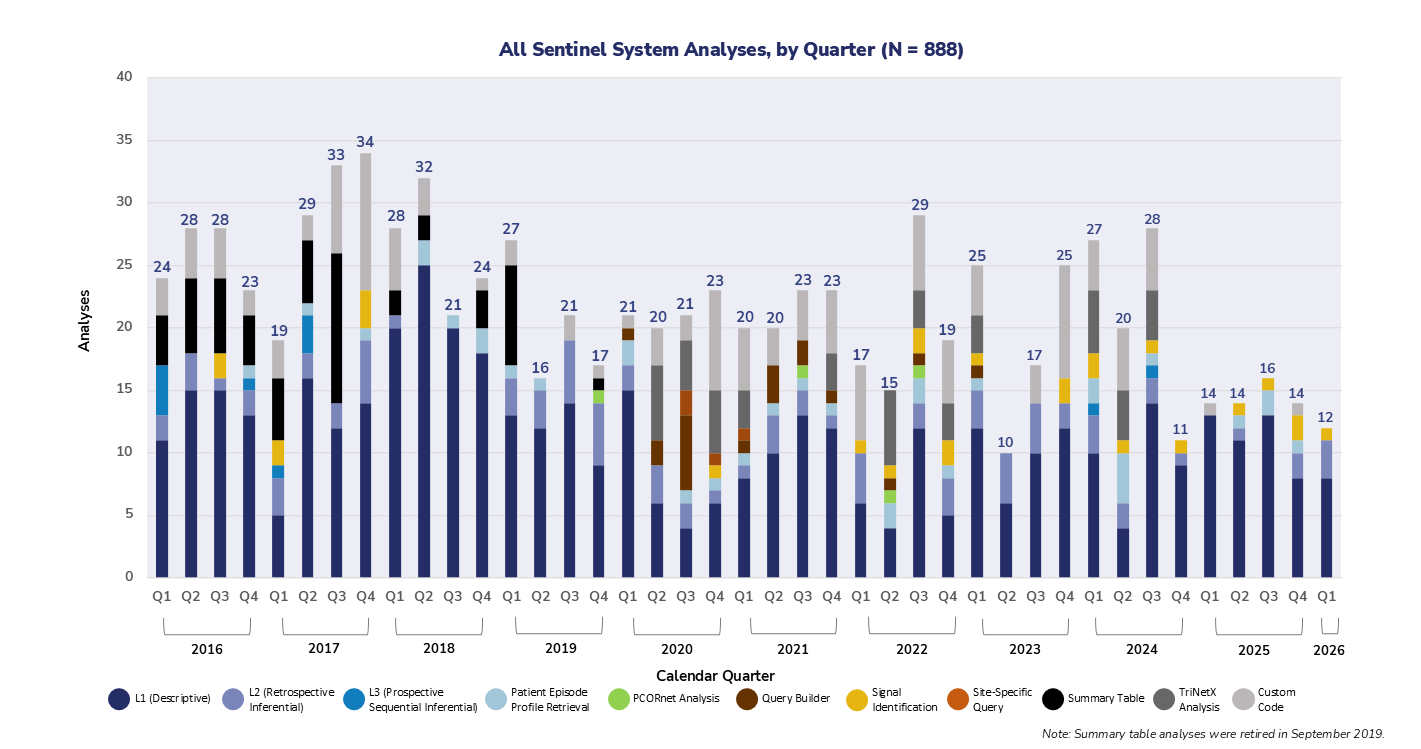
The graph below captures the total number of analyses conducted by the FDA since 2016 throughout the Sentinel System, including the Sentinel Distributed Database (SDD), IBM Watson Health, IBM Explorys, TriNetX, HCA Healthcare, PCORnet, and Veradigm.
Sentinel’s ARIA system is complemented by EHR data systems, enabling the selection of a data source that best fits a drug safety question of interest. There are 3 categories of EHR data sources:
Data Aggregators: Compiles EHR data from multiple, discrete healthcare organizations in a single platform.
Data Warehouse: Stores extracted, standardized data from transactional systems in a central repository.
Network: An integrated partnership of standardized EHR data among multiple data partners.
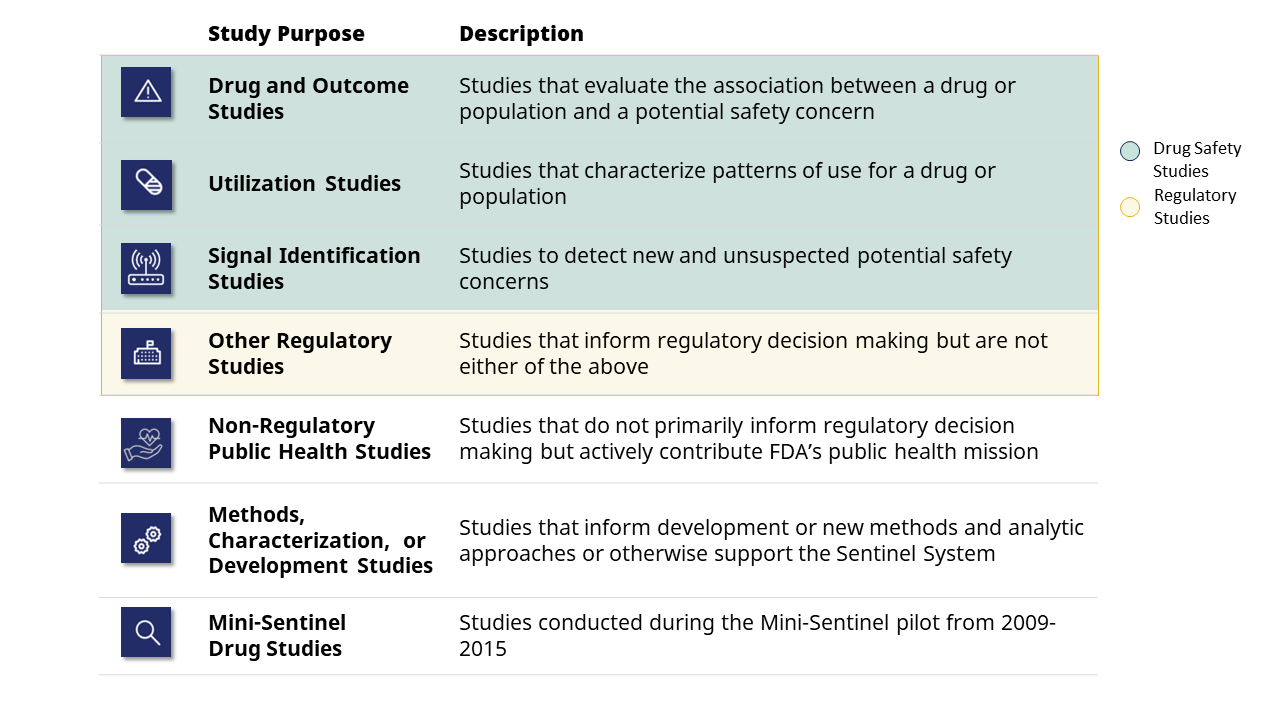
All drug safety studies conducted in Sentinel are included below. The table is organized by the drug or population of interest and the health outcome(s) under study. When available, links are provided to the analytic code (package), results, communications and the study’s regulatory outcome.
The U.S. Food and Drug Administration (FDA) initiated this study in the Sentinel System to evaluate a potential association between use of glucagon-like peptide‑1 receptor agonists (GLP‑1 RAs) and intentional self-harm. FDA began investigating an association between GLP‑1 RAs and suicidal ideation and behavior (SI/B) in July 2023 upon learning of post-marketing reports of (SI/B) in patients taking GLP-1 RAs. FDA performed a preliminary review of clinical trials and postmarketing data, including observational studies and case reports, and publicly reported those findings in its January 2024 Drug Safety Communication. FDA conducted analyses using U.S. claims data to assess the risk of intentional self-harm following new use of GLP-1 RAs, in the Sentinel Distributed Database, to further evaluate the safety signal. FDA conducted both comparative and descriptive analyses with Sentinel System data.
In comparative analyses, the primary analyses compared the risk of intentional self-harm between new users of GLP-1 RA and sodium-glucose cotransporter-2 (SGLT-2) inhibitors, and separately between GLP-1 RA and dipeptidyl peptidase-4 (DPP-4) inhibitors, in a large population of commercially and privately insured individuals aged 18 years or older with type 2 diabetes. The adjusted incidence rates of intentional self-harm per 1,000 person‑years were 1.13 events for GLP‑1 RAs, 1.22 events for SGLT‑2 inhibitors, and 1.37 events for DPP‑4 inhibitors. Adjusted hazard ratios (HR) did not show an increased risk among GLP‑1 RA users compared with either comparator (GLP‑1 RAs vs. SGLT‑2 inhibitors: HR 0.93; 95% CI 0.81, 1.08; GLP‑1 RAs vs DPP‑4 inhibitors: HR 0.94; 95% CI 0.82, 1.07). Similarly, FDA did not find an increased risk in the subgroup of patients with both type 2 diabetes mellitus and obesity.
FDA also conducted a descriptive analysis among patients aged 18 years or older with obesity and without evidence of diabetes or pregnancy to evaluate the occurrence of intentional self-harm following incident use of GLP‑1 RAs or other weight‑loss agents.
Results from the Sentinel comparative analyses supported FDA’s investigation of the safety concern and informed FDA’s evaluation of GLP-1 RAs and SI/B. Considering the totality of the evidence, including results from the Sentinel analyses, an internal meta-analysis of clinical trials across GLP-1 RA drug development programs, as well as a review of published studies, FDA issued a Drug Safety Communication summarizing its findings of no increased risk of SI/B associated with the use of GLP-1 RA medications. FDA requested removal of the warning for suicidal behavior and ideation from the labeling of Saxenda (liraglutide), Wegovy (semaglutide), and Zepbound (tirzepatide) on January 13, 2026.
The U.S. Food and Drug Administration (FDA) initiated this study in the Sentinel System to better understand genetic testing patterns for dihydropyrimidine dehydrogenase (DPYD) gene variants among patients treated with fluoropyrimidines in the United States.
The enzyme dihydropyrimidine dehydrogenase, encoded by the DPYD gene, plays an important role in the breakdown of fluoropyrimidines. Fluoropyrimidines are widely used for the treatment of solid tumors. Individuals with certain homozygous or compound heterozygous variants of the DPYD gene have reduced enzyme activity and are at increased risk for acute early-onset toxicity and serious adverse reactions. Warnings related to DPD deficiency for fluoropyrimidine products were incrementally strengthened through multiple labeling updates since 2015. Most recently, the labeling for Xeloda (capecitabine) and fluorouracil products was updated in October 2025 and February 2026, respectively, to recommend DPYD testing before fluoropyrimidine administration. FDA used the Sentinel Distributed Database to assess the frequency of DPYD gene testing among insured patients initiating fluoropyrimidine treatment in the United States, in the period before the most recent updated product labeling.
The results indicated DPYD testing is infrequent among insured patients receiving fluoropyrimidine treatment. During the study period, January 1, 2018, to May 31, 2025, DPYD testing (i.e., testing within ± 60 days of treatment initiation) was performed in 0.4% of patients who received any fluoropyrimidine overall, in 0.3% of patients who received testing in the 60 days before initiating any fluoropyrimidine treatment, and in 0.2% of patients who received DPYD testing within 60 days after initiating treatment for any fluoropyrimidine.
The results helped FDA to understand DPYD testing patterns before and after fluoropyrimidine administration. Recent revisions to fluoropyrimidine product labeling included recommendations to test patients for DPYD variants before treatment. These results will allow FDA to assess whether and how testing patterns shift in response to those labeling changes.
The U.S. Food and Drug Administration (FDA) initiated this study to explore the feasibility of using real-world setting data (TriNetX Live™ platform) to characterize patients undergoing allogeneic hematopoietic stem cell transplantation (allo-HSCT) and to evaluate the characteristics and adverse outcomes of patients who experience transplant associated thrombotic microangiopathy (TA-TMA) in the pediatric and adult populations. An additional objective of this study was to characterize the ability of TriNetX to capture clinical data necessary to determine whether TA-TMA cases are considered to be high risk.
Between July 1, 2009, and August 31, 2020, 12,350 adult patients (aged 18 years and older) and 1,300 pediatric patients (aged younger than 18 years) with allo-HSCT were observed. TMA occurring within 12 months of allo-HSCT was observed among 290 (2.3%) adults and 60 (4.6%) pediatric patients, with mean ages (SD) at TMA diagnosis of 39.2 (21.1) years and 3.1 (2.67) years, respectively. Among patients with allo-HSCT, mortality within 12 months following transplant was observed in 3,160 (25.6%) adults and 180 (13.8%) pediatric patients. Among patients with TA-TMA, mortality within 12 months following TMA diagnosis was higher in both populations: 130 (44.8%) in adults and 20 (33.3%) in pediatric patients.
The study findings informed the FDA about the possibility of identifying potential external controls for clinical programs. The study findings also facilitated the understanding of the characteristics and outcomes of allogeneic HSCT population, particularly among the TA-TMA pediatric and adult patients who are identified using diagnostic codes.
The Coronavirus Disease 2019 (COVID-19) was first detected in the United States in January 2020. Early studies indicated a disproportionate burden of COVID-19 infections, hospitalizations, and deaths among racial and ethnic minority groups. Importantly, these early studies were limited in geographic scope, and/or focused only on elderly and higher-risk adults. The COVID-19 pandemic, thus, presented the FDA with a unique opportunity to evaluate race and ethnicity-related data pertaining to COVID-19 testing, hospitalizations, and other outcomes, including disease severity and mortality. FDA’s studies in the Sentinel System sought to expand upon the current understanding of COVID-19-related outcomes among insured adult patients under 65 years old by geographic region over the year-long study period, using Sentinel’s rapid Sentinel Distributed Database (SDD), TriNetX Network, and PCORnet data sources.
The rapid SDD analysis found up to 42% of insured individuals with COVID-19 had missing race information, and patients with unknown race were younger and healthier. Results also indicated that minoritized individuals with COVID-19 have increased odds of COVID-19 hospitalization, critical COVID-19, and 30-day in-hospital mortality after controlling for baseline differences.
An analysis conducted in the PCORnet Distributed Data Network identified approximately 580,000 patients with any evidence of COVID-19 and approximately 73,000 with evidence of inpatient COVID-19 between April 2020 and March 2021. Data on race and ethnicity were well-populated in this electronic health record data source, especially compared to claims data sources (<9% of all eligible patients had missing race data). While there were no statistically significant differences in the racial composition of the "all eligible" and "any COVID-19" cohorts, patients of "Black or African American" and "Other" race were disproportionately present in the hospitalized COVID-19 cohort (21.2% vs 14.3% for Black and 9.3 vs 5.9% for Other). Apparent differences in clinical observation and laboratory results existed between racial/ethnic minoritized groups compared to patients of White race and/or Non-Hispanic ethnicity, but interpretation of these findings was difficult due to lack of standardization. Future work should examine to what extent clinical and laboratory data can be used to understand potential racial and ethnic differences in COVID-19.
This study raised awareness of the extent of missingness of race and ethnicity in administrative claims data used for regulatory purposes. This evaluation of missing race and ethnicity data provoked innovative thinking of approaches to improve the capture of race data in the Sentinel Distributed Database. Building a full picture of the associations between race and ethnicity and COVID-19 outcomes will continue efforts to characterize the full impact of the COVID-19 pandemic. As this study intended to characterize the burden of COVID-19 outcomes, FDA did not take any regulatory action based on these data.
This project was funded by U.S. Food and Drug Administration (FDA) Office of Minority Health and Health Equity (OMHHE).
Ketamine is a rapid-acting general anesthetic indicated for: (a) anesthesia for diagnostic and surgical procedures not requiring skeletal muscle relaxation, (b) induction of anesthesia prior to other general anesthetic agents, and (c) supplementation of other anesthetic agents. In August 2020, the Substance Abuse and Mental Health Administration (SAMHSA) inquired about the off-label use of ketamine for agitation by Emergency Medical Services (EMS) and in the emergency department (ED). In response, the U.S. Food and Drug Administration (FDA) initiated this study in the Sentinel System to understand the use of ketamine in EDs, by EMS, and in other pre-hospital settings, particularly for the treatment of agitation.
This study utilized the TriNetX USA Network Database, a Sentinel collaborator, which contained electronic medical records (EMR) from 67 healthcare organizations for approximately 90 million patients as of June 2020. The study population included unique patients with mental health diagnoses on the same day as, or the day following, an ED visit between January 1, 2016, and June 30, 2020. The study identified some use of ketamine in the defined population. In 2019, among 363,530 patients with a mental health diagnosis related to acute agitation on the day of an ED visit, less than 1% received ketamine (n=2,689 received any ketamine; n=865 received ketamine without a benzodiazepine or antipsychotic drug). While the available data suggested that ketamine may have been administered to help manage agitation, specific clinical reasons were difficult to determine due to data limitations.
This study supported the FDA’s broader efforts to monitor real-world drug utilization. Based on these Sentinel study findings, and information from other sources, no regulatory actions were recommended.
The U.S. Food and Drug Administration (FDA) initiated this study in the Sentinel System to estimate the prevalence of cutaneous lupus erythematosus (CLE) and its subtypes. Existing prevalence estimates were limited, and CLE includes several subtypes that differ significantly, making it challenging to understand the true burden of disease.
This analysis assessed the prevalence of CLE subtypes overall, by diagnosing physician specialty, and across demographic groups, including sex, race, ethnicity, and insurance type.
Results showed that CLE subtypes are rare (less than 100 per 100,000 eligible members) with an overall low prevalence in the Sentinel Distributed Database (SDD). Among dermatologist- or rheumatologist-confirmed cases, the prevalence of discoid lupus erythematosus (DLE) was 27.77 per 100,000 eligible members and 9.28 per 100,000 eligible member-years (95% Confidence interval [CI]: 9.21, 9.35). Subacute CLE (SCLE) was 9.81 per 100,000 eligible members and 3.28 per 100,000 eligible member-years (95% CI: 3.24, 3.32). Other subtypes included: other cutaneous lupus erythematosus (OLE) at 11.71 per 100,000 members and 3.91 per 100,000 member-years (95% CI: 3.87, 3.96); systemic lupus erythematosus without systemic manifestations at 14.38 per 100,000 members and 4.81 per 100,000 member-years (95% CI: 4.76, 4.85); DLE preceding systemic lupus erythematosus at 11.22 per 100,000 members and 3.75 per 100,000 member-years (95% CI: 3.71, 3.79); mixed CLE at 11.40 per 100,000 members and 3.81 per 100,000 member-years (95% CI: 3.77, 3.85); and Chilblains with evidence of systemic lupus erythematosus at 0.67 per 100,000 members and 0.22 per 100,000 member-years (95% CI: 0.21, 0.23).Prevalence of all CLE subtypes was higher among women than men, and greatest in adults aged 45 to 64 years with an extremely low prevalence in patients younger than 18 years of age. Black and White patients had the highest prevalence, and non-Hispanic individuals had higher prevalence than Hispanic individuals. Prevalence of CLE subtypes differed between publicly and commercially insured patients, and differences persisted across race and sex strata.
These findings will support ongoing FDA reviews. Based on the study findings, no immediate regulatory actions were required, but the results fill critical knowledge gaps and strengthen the scientific foundation for future decision making.
The U.S. Food and Drug Administration (FDA) conducted this study to expand previous work examining the safety and effectiveness of non-vitamin K antagonist oral anticoagulants (NOAC) for non-valvular atrial fibrillation (NVAF) in users aged 65 years and older. In a previous FDA study using US Medicare data, dabigatran and apixaban had a more favorable benefit−harm profile compared to rivaroxaban in users aged 65 years and older. It was decided to examine whether a similar benefit-harm profile persists in younger users. Before conducting the current study, findings from the Medicare study were replicated in patients aged 65 years and older in the Sentinel System (Medicare only) to determine study feasibility and to assess if findings were comparable across data sources and analytic approaches. Specifically, the current study examined the comparative effectiveness and safety of NOACs examining risk of bleeding (major extracranial bleeding (MEB), gastrointestinal bleeding (GIB), intracranial hemorrhage) and thromboembolic stroke in NOAC users with NVAF aged under 65 years.
A total of more than 173,000 patients (mean [SD] age, 56.6 [7.23] years; 27.5% female; 72.5% male) were included across the 3 exposure cohorts. The number of NOAC users for each pairwise comparison were rivaroxaban (57 932) vs apixaban (96 057), rivaroxaban (57 399) vs dabigatran (20 188), and dabigatran (20 163) vs apixaban (96 668). Rivaroxaban was associated with higher risks of MEB and GIB compared with apixaban (MEB: HR, 1.91; 95% CI, 1.56-2.34; GIB: HR, 1.92, 95% CI, 1.54-2.39), while differences in thromboembolic stroke prevention were not significant (HR, 1.05; 95% CI, 0.77-1.44). Dabigatran was associated with higher thromboembolic stroke risk (rivaroxaban vs dabigatran: HR, 0.61; 95% CI, 0.39-0.94; dabigatran vs apixaban: HR, 1.74; 95% CI, 1.13-2.68)- a finding that was not observed in the previous FDA study of patients aged 65 years and older.
These findings suggest that for patients aged younger than 65 years treated with NOACs for NVAF, apixaban was associated with the most favorable benefit-harm profile. While the results suggest that rivaroxaban may have provided greater stroke prevention than dabigatran, it was associated with higher bleeding risks than apixaban without additional stroke prevention. The higher thromboembolic stroke risks observed with dabigatran in younger patients suggest important age-related differences in effectiveness that warrant further investigation. Based on the study findings, FDA determined no regulatory actions were required.
Pharmacoepidemiologic studies based on administrative claims data are sometimes limited in their ability to control for confounding, either due to unavailability of certain clinical information, poor accuracy, and/or incompleteness of coded information. The capture of confounding covariates in U.S. administrative claims data may be improving over time, but little empirical data are available to support this statement. Electronic health records (EHRs) include more detailed information on patients’ care that is not routinely available in administrative claim databases.
The U.S. Food and Drug Administration (FDA) initiated these studies in the Sentinel System to examine the capture of key confounding covariates in electronic healthcare databases (administrative claims and EHR) to support regulatory drug safety evaluation. Covariates of interest from the Sentinel Distributed Database (SDD) included in the analyses were smoking, obesity, overweight, body mass index (BMI), alcohol abuse/dependence, drug abuse/dependence, systolic/diastolic blood pressure (BP), reported race, history of Coronary Artery Bypass Graft (CABG) or Percutaneous Transluminal Coronary Angioplasty (PTCA), and history of sudden cardiac arrest. Covariates included from the TriNetX U.S.A. Network (EHR data) were obesity, smoking, BMI, and systolic/diastolic BP.
The findings from SDD analyses using administrative claims data spanning 2006 to 2023 showed that the capture of most of these common confounding covariates improved over time with the lowest to highest prevalent values during this period being 1.9% to 18.4% for obesity, 0.2% to 9.2% for overweight, 2.3% to 14.8% for smoking, 0.4% to 2.0% for alcohol abuse/dependence, 0.2% to 1.8% for drug abuse/dependence, 0.6% to 3.8% for history of CABG or PTCA, and 0% to 0.4% for history of sudden cardiac arrest. However, a low prevalence of some of the covariates compared to national estimates in 2023 suggests that these covariates remain inadequately documented in U.S. claims data (obesity 18.4% vs 34.3%, overweight 9.2% vs 34.4%, alcohol abuse/dependence 2.0% vs 6.1%, and drug abuse/dependence 1.8% vs 17.1%, respectively).1
The analysis conducted in aggregated EHR data from TriNetX also showed a lower prevalence of obesity (14.0%) when compared with national estimates (34.3%), while the prevalence of smoking captured in both SDD (14.8%) and TriNetX (12.0%) was higher than or similar to the national estimate (12.1%). Systolic/diastolic BP and BMI were captured in about 50-60% of eligible members in TriNetX.
These exploratory Sentinel studies provided information to FDA on the availability of common confounders in electronic healthcare databases to aid decisions on the use of Sentinel data in future drug safety studies. This work highlighted the importance of carefully evaluating each data source to determine whether it can adequately address the specific study question by capturing the key confounding covariates required for the analysis. It also suggests a need to consider alternative approaches such as quantitative bias analysis in the early phase of study design when confounders are poorly captured, to assess potential bias from unmeasured confounding.
1National estimates from CDC Behavioral Risk Factor Surveillance System (BRFSS) and Substance Abuse and Mental Health Services Administration (SAMHSA) National Survey on Drug Use and Health.
The U.S. Food and Drug Administration (FDA) initiated this study in the Sentinel System to assess the risk of serious infection in patients with Crohn’s disease (CD) or ulcerative colitis (UC) during the use of ustekinumab (Stelara). The study was prompted by uncertainties that arose during the drug’s review regarding the magnitude or nature of risk posed by ustekinumab on immune function, as well as limited information available from clinical trials at the time. The primary goal of this analysis was to exclude two-fold or greater risk of serious infection in adult CD patients using ustekinumab when compared to adult CD patients using a biologic treatment other than ustekinumab.
Sentinel estimated incidence at 44.1 and 51.6 patients with at least one serious infection or COVID-19 event per 1,000 patient-years of treatment with ustekinumab and comparator biologic, respectively. Sentinel estimated risk with hazard ratio (HR) 0.88 and 95% confidence interval (CI) 0.80-0.96. Based on these results, FDA determined that the safety information in ustekinumab labeling related to the risk of serious infection in patients with CD was adequate.
FDA continues to evaluate the risk of serious infection in patients with UC following use of ustekinumab. Updates will be provided as information becomes available.
A single dose of doxycycline can serve as effective post-exposure prophylaxis for Lyme disease following a high-risk tick bite. Despite historical concerns about doxycycline toxicity in children under eight years of age, recent guidelines state that treatment courses of less than 21 days are safe for children of any age.
To build upon earlier research focused primarily on adults, the Centers for Disease Control and Prevention (CDC) collaborated with the U.S. Food and Drug Administration (FDA) to leverage the Sentinel System's comprehensive data, including commercial insurance and Medicaid claims, to provide new insights into doxycycline dispensing for both adult and pediatric populations.
The study aimed to characterize the patterns of single-dose doxycycline dispensing by:
Describing state-level trends in pediatric and adult prescription claims.
Examining patient-level factors such as age, sex, race, ethnicity, and urbanization in pediatric and adult prescription claims.
Quantifying how many pediatric and adult patients received multiple courses of single-dose doxycycline.
Comparing dispensing trends in pediatrics between Medicaid and commercial insurance.
Results indicated dispensing patterns of single-dose doxycycline in both adults and children closely aligned with known Lyme disease epidemiology, showing higher rates in the Northeast and Mid-Atlantic regions and seasonal peaks in spring, summer, and fall. Importantly, the findings also suggested that Lyme disease post-exposure prophylaxis may be underutilized in the pediatric population, particularly among children covered by Medicaid.
The findings from the Sentinel analyses supported CDC’s ongoing Lyme disease surveillance efforts and contributed important insights that build on CDC’s previous work by providing valuable data on post-exposure prophylaxis use among the pediatric population.
Hypoglycemia is a well-known adverse reaction associated with beta blocker use. During routine post-market surveillance, the FDA identified a case report describing severe hypoglycemia in an infant associated with nadolol therapy for infantile hemangioma. While select beta blockers are approved by the FDA for specific pediatric indications including hypertension (metoprolol succinate), hemangioma (Hemangeol, an oral propranolol solution), and lowering of intraocular pressure (timolol ophthalmic solution), various beta blockers may be used for a range of off-label conditions in this population. The hypoglycemia safety signal and the limited understanding of the magnitude and patterns of beta-blocker use in the pediatric population prompted a Sentinel study to comprehensively characterize beta blocker use and assess the incidence of hypoglycemia associated with various beta blockers in this population.
Findings indicated that beta blockers were used for a variety of cardiac and non-cardiac conditions in the pediatric population, with potential indications for use varying by age groups. Notably, the study found that beta blocker use for indications approved by the FDA (Hemangeol for hemangioma and metoprolol succinate for hypertension) accounted for only 2% of all beta blocker use in the pediatric population. Additionally, children five years of age and younger appeared to be more prone to experience hypoglycemia following beta-blocker initiation.
Data from the FDA Adverse Event Reporting System, published case reports, the National Poison Data System, and this Sentinel study suggest that the association between beta blocker exposure and hypoglycemia in the pediatric population was observed with products across the beta blocker class. Based on the totality of data, the FDA approved safety-related labeling changes for beta blocker products to describe the risk of hypoglycemia in pediatrics or individuals unable to communicate signs of hypoglycemia. These changes are captured in the “Warnings and Precautions” and “Patient Counseling Information” sections of the Prescribing Information.
Pregnant women are rarely included in clinical trials. Yet, pregnant women may choose to continue medical treatment during pregnancy, particularly for chronic conditions. Therefore, FDA’s capacity to study medical product exposures during pregnancy on maternal and infant outcomes is important to monitor the safety of medication use during pregnancy. Pregnancy registries or postmarket observational studies typically are used to evaluate the safety of medication use during pregnancy.
The purpose of this study was to characterize the demographics, health characteristics, and drug utilization of infants and mothers included in the Sentinel Distributed Database (SDD) Mother-Infant Linkage (MIL) table. The study additionally characterized infants and pregnant females not included in the MIL table. These were infants who had not been linked to a mother, mothers with a documented singleton birth but without linkage to her infant and pregnant females with a non-live birth outcome.
For the infant component, using data from the MIL table for both Medicaid and non-Medicaid data sources, we characterized about 5.4 million infants with matched mothers and 17.4 million without matches. The maternal component provides extensive data on comorbidities, drug use, and pregnancy characteristics for more than 6.7 million live birth and 3.2 non-live birth deliveries. No regulatory outcomes were intended for this study as it was designed for purposes of describing the infant and maternal groups included in the SDD MIL table.
The U.S. Food and Drug Administration (FDA) identified a potential safety signal for anaphylaxis and angioedema with the use of Keppra (levetiracetam) during routine post-market safety surveillance of the FDA Adverse Event Reporting System (FAERS). To evaluate this signal, the FDA conducted an integrated safety review that included an analysis in the Sentinel System, along with a review of FAERS cases, other data sources, and medical literature.
The Sentinel analysis compared the incidence of angioedema among new users of levetiracetam to that of new users of other antiepileptic drugs. The analysis found that the incidence rate of angioedema in new users of levetiracetam was similar to the rates for other antiepileptic drugs that were already labeled for this risk. Based on the totality of evidence from the case series and the Sentinel analysis, the FDA approved a safety-related labeling change for Keppra (levetiracetam) on April 24, 2017, to add the risk of anaphylaxis and angioedema to several sections of the label, including the Warnings and Precautions section.
The U.S. Food and Drug Administration (FDA) initiated this study in the Sentinel System to characterize the utilization of ingestible sodium fluoride drug products among pediatric Medicaid patients, in preparation for a public meeting held to discuss the use, benefits, risks, and evidence gaps related to ingestible sodium fluoride drug products. The study assessed the amount of dispensing for orally ingestible sodium fluoride drug products among both pediatric and adult Medicaid patients, as well as the distribution of product National Drug Codes (NDCs) dispensed to Medicaid patients in the Sentinel System. The study also assessed the amount of dispensing for ingestible dietary supplements that contain sodium fluoride for background context.
Results showed that from 2018 to 2021, the use of ingestible sodium fluoride drug products decreased over time among the various pediatric age groups and that these products were most frequently dispensed to pediatric patients aged three to nine years. The analysis identified 21 ingestible sodium fluoride drug product NDCs dispensed during the study period among the Sentinel Medicaid patients.
The study provided information on how ingestible sodium fluoride drug products are used among pediatric patients in the Medicaid population, which represents a subset of the broader U.S. population exposed to ingestible sodium fluoride drug products. The Sentinel Medicaid analysis results supported FDA’s preparation for the public meeting.
Faricimab-svoa is a bispecific antibody currently approved for treatment of neovascular (wet) age-related macular degeneration (nAMD), diabetic macular edema (DME), and macular edema following retinal vein occlusion (RVO). Faricimab-svoa was selected for this signal identification study based on high utilization and its status as a recently approved new molecular entity.
The U.S. Food and Drug Administration (FDA) initiated one analysis in Sentinel using a tree-based scan statistic to support this safety signal identification for faricimab-svoa. A self-controlled risk interval design with Tree-Only Scan and Tree-Temporal Scan methodologies were used to evaluate incident outcomes and temporal clusters of incident outcomes, respectively, among new users of faricimab-svoa.
The goals of the study were (1) to monitor non-pregnancy and non-cancer safety outcomes among new users of faricimab-svoa; (2) to identify any imbalances in safety outcomes after treatment initiation compared with a baseline time period before treatment initiation, and (3) evaluate how such safety outcomes are distributed in time after treatment initiation. Overall, the study did not identify any new safety concerns for faricimab-svoa in this sample of real-world patients.
Statistical alerts are triaged in consideration of study design, existing drug knowledge, therapeutic context, treated population, and potential public health impact. Alerts determined to be newly identified safety signals (NISS) follow the FDA’s Center for Drug Evaluation and Research manual of policies and procedures for NISS (MAPP 4121.3). After evaluating the statistical alerts generated by this study in the context of other information, the FDA did not identify any new safety concerns and determined that no further evaluation is warranted at this time.
Sotagliflozin is a sodium-glucose cotransporter-2 (SGLT2) inhibitor. Data from the Zynquista (sotagliflozin) clinical studies reported an increased risk of diabetic ketoacidosis (DKA) in patients with type I diabetes mellitus (T1DM) exposed to sotagliflozin compared to the placebo arm. In 2018, to support the review of Zynquista (sotagliflozin), proposed for T1DM, the U.S. Food and Drug Administration (FDA) initiated a study using the FDA Sentinel System to describe the characteristics and risk of DKA among patients with T1DM who received SGLT2 inhibitors from 2013 to 2018. The 2018 study found that the observed rates of DKA among patients with T1DM exceeded the expectation based on the Zynquista clinical trials. These findings were presented in the January 17, 2019 Endocrinologic and Metabolic Drugs Advisory Committee (EMDAC) meeting, and informed the discussion on the benefit-risk assessment of Zynquista.
In 2024, to support the review of Zynquista (sotagliflozin), proposed for T1DM with chronic kidney disease (CKD), FDA investigated the occurrence of DKA (1) in patients with T1DM who initiated SGLT2 inhibitors to inform the DKA risk over time and (2) in patients with T1DM by CKD stages to describe risk of DKA in patients with T1DM at different stages of CKD, using the Sentinel Distributed Database. Findings from these descriptive analyses observed that crude incidence rates of DKA in patients with T1DM who initiated SGLT2 inhibitors did not appear to have declined from 2013 to 2024 and that there is an increase in incidence rate of DKA with advancing CKD stage in patients with T1DM, regardless of SGLT2 inhibitor use. These findings were presented in the October 31, 2024 EMDAC meeting, where the benefits and risks of sotagliflozin as an adjunct to insulin therapy to improve glycemic control in adult patients with T1DM with CKD were discussed.
FDA also conducted a comparative analysis to assess the risk of DKA by CKD stage in patients with T1DM. Compared to patients with T1DM and CKD stage 1 or 2, the crude incidence rates of DKA were significantly higher in patients with T1DM and CKD stage 3 or CKD stage 4 or 5. Though FDA conducted an additional analysis using 1:1 propensity score matching to balance the baseline characteristics of patients with T1DM at different stages of CKD, there were limitations in interpretation of findings with covariate adjustments. There was limited overlap in propensity score for the respective comparison groups (i.e., 56% of subjects with advanced CKD were excluded from analyses because of non-overlapping propensity score), and comparative analyses results were applicable to a subset of patients with T1DM with advanced CKD. Furthermore, given that CKD is strongly associated with increased patient age, duration of diabetes, and comorbidities that contribute to progression of CKD, such as hypertension, it may not be clinically meaningful to estimate the risk of DKA under the assumption that patients with and without advanced CKD had similar characteristics in terms of age and certain comorbidities (e.g., hypertension). The results of the descriptive analyses (i.e., the crude incidence rates) helped inform FDA’s understanding of DKA in patients with T1DM and CKD.
Under PDUFA VII Pregnancy Safety Commitments, FDA will develop a framework describing how different types of post-market studies might optimally be used to assess pregnancy safety. The framework considers factors such as, but not limited to, purpose of study, types of post-market studies, anticipated exposure in females of reproductive potential (FRP) and pregnant women, potential toxicity of the drug and proposed risk mitigation, benefits of the drug, and magnitude and type of risk to be detected. To inform the development of this framework, it is important to understand how different types of post-market studies have been used by FDA and identify knowledge gaps.
This study examined the sources and characteristics of quantitative human pregnancy safety data contained in the “Human Data” subsection of Section 8.1 for products with “Pregnancy and Lactation Labeling Rule” (PLLR)-converted drug labeling during the period June 30, 2015, to December 31, 2021. As part of PDUFA VII Pregnancy Safety Commitments, this study aimed to conduct a review of the types of post-market human pregnancy data that have been included in pregnancy labeling. The study found that quantitative data included in the pregnancy subsection 8.1 was not limited to any particular type of study design. The Sentinel Distributed Database was used to assess the utilization during pregnancy of the products with PLLR-converted drug labeling changes related to pregnancy from January 1, 2008, to September 30, 2023. This analysis showed that study types incorporated in labeling did not always correlate with prevalence in use of products during pregnancy.
The U.S. Food and Drug Administration (FDA) initiated this study in the Sentinel System to assess the cardiovascular safety of tirzepatide. Results from the placebo-controlled clinical trials for Mounjaro (tirzepatide) reported a dose-related heart rate increase in the tirzepatide-treated subjects, with higher frequencies reported in trials conducted in Japan. The FDA conducted this descriptive analysis in a real-world setting to assess the clinical relevance of the clinical trial findings and inform future Sentinel assessments as needed.
This Sentinel study sought to describe the incidence rate of hospitalized tachyarrhythmia in patients with type 2 diabetes who newly initiated Mounjaro or other long-acting glucagon-like peptide-1 receptor agonists (GLP-1 RAs). Other GLP-1 RAs included in the analysis were: Bydureon (exenatide), Bydureon BCISE (exenatide), Ozempic (semaglutide), Rybelsus (semaglutide), Wegovy (semaglutide), Saxenda (liraglutide), Victoza (liraglutide), Trulicity (dulaglutide), and Xultophy (insulin degludec/liraglutide). Hospitalized tachyarrhythmia was defined as the presence of any of the following conditions: atrial fibrillation and flutter, sinus tachycardia, supraventricular tachycardia, other tachycardia, cardiac arrest, ventricular fibrillation and flutter, and ventricular tachycardia.
The results described the incidence rate of hospitalized tachyarrhythmia by product and patient characteristics, including race subgroups. From May 13, 2022, to June 30, 2023, the study identified 18,842 new users of Mounjaro. Of these, 102 (0.54%) experienced hospitalized tachyarrhythmia, resulting in an incidence rate of 472.43 per 10,000 patient-years. Among the 108 Asian new users of Mounjaro, none experienced an event of hospitalized tachyarrhythmia. Among other GLP-1 RAs, Ozempic had the largest number of new users (N= 117,008), followed by Trulicity (N= 66,978) and Rybelsus (N= 34,058). The incidence rate of hospitalized tachyarrhythmia in the eight GLP-1 RA products was 392.98 per 10,000 patient-years for Bydureon, 488.45 per 10,000 patient-years for Ozempic, 381.28 per 10,000 patient-years for Rybelsus, 199.10 per 10,000 patient-years for Saxenda, 500.90 per 10,000 patient-years for Trulicity, 137.10 per 10,000 patient-years for Wegovy, 601.14 per 10,000 patient-years for Victoza, and 349.71 per 10,000 patient-years for Xultophy.
The results of this Sentinel study contributed to the FDA's discussion regarding the planned pharmacovigilance activities for tirzepatide. Due to the descriptive nature and the short evaluation period, this study did not provide definitive information regarding the cardiovascular safety of tirzepatide. The FDA concluded that an additional Sentinel analysis was not warranted since no safety signals were identified through other pharmacovigilance activities thus far, no major findings emerged from related studies for tirzepatide, and data from the cardiovascular outcomes trial (CVOT) for Mounjaro are anticipated to be available by the end of 2025.
The U.S. Food and Drug Administration (FDA) initiated this study in the Sentinel System to evaluate potential rebound of multiple sclerosis (MS) disease activity following discontinuation of fingolimod hydrochloride, particularly in the context of pregnancy planning. Fingolimod hydrochloride is used to treat relapsing forms of MS, a chronic autoimmune condition characterized by episodic neurological relapses. While MS therapies are effective in reducing relapse rates and delaying progression, discontinuation—especially when abrupt—has been associated with a rebound phenomenon, where disease activity returns in a more severe form.
This feasibility analysis specifically aimed to estimate MS relapse frequency and rate as the primary indicators of disease activity, and to compare the performance of MS relapse algorithms found in the literature. We assessed four cohorts: prevalent MS-fingolimod hydrochloride-exposed, incident MS-fingolimod hydrochloride-exposed, prevalent MS-fingolimod hydrochloride-never exposed, and incident MS-fingolimod hydrochloride-never exposed. Five algorithms were used to identify MS relapse. Between September 21, 2010, to January 31, 2018, the total number of patients exposed to fingolimod hydrochloride was 7,589 prevalent MS patients and 1,039 incident MS patients. The total number of fingolimod hydrochloride-never exposed members was 216,429 prevalent MS patients and 133,567 incident MS patients. MS relapse rates varied by algorithm, such that algorithms that included diagnoses in outpatient settings captured more MS relapses than algorithms that included diagnoses in inpatient settings alone. The findings were intended to inform regulatory decision-making and clinical guidance on the safe discontinuation of fingolimod, particularly for women planning pregnancy where interruption of treatment may be necessary.
Ultimately, the FDA did not pursue further analyses, as a related regulatory action—label change to add “severe increase in disability after stopping GILENYA” to the Warnings and Precautions—had been taken.
The U.S. Food and Drug Administration (FDA) initiated this study in the Sentinel System to determine the patterns of congenital cytomegalovirus infection (cCMV) diagnosis, trends in prescribing valganciclovir (vGCV) for treatment of cCMV and associated clinical outcomes (e.g. hearing and hematologic) for infants with cCMV. Congenital cytomegalovirus infection (cCMV), a rare pediatric disease, may be associated with serious audiologic and neurodevelopmental impairment. Although there are currently no FDA-approved agents to prevent or treat cCMV infection, clinical guidelines recommend vGCV treatment for six months, initiated within the first month of life in newborns with moderate to severe disease. It is unknown to what extent the off-label vGCV use for cCMV has become the standard of care. This knowledge gap impacts clinical development of antivirals for treatment of cCMV.
In a large cohort of infants (n=1500) with cCMV identified by claims data, 20% were treated with vGCV. Of the treated population, 17% were asymptomatic, 27% commenced treatment outside the first month of life, and 38% received treatment for longer than 6 months. Although clinical severity cannot be determined from claims data, these results suggest that vGCV treatment in the U.S. may extend beyond the current recommendation. Severe hematological events occurred infrequently. The proportion of infants with cCMV infection who had hearing loss increased over time, regardless of treatment. However, a subsequent analysis of patient charts suggested that only a subset (45%) of the children identified using claims data had confirmed cCMV infection, which limits the interpretability of results from that inquiry.
This Sentinel Study was also used to examine the availability of real-world data (RWD) to complement clinical trial data for regulatory decision-making. Results were reviewed for insights on the use of RWD to construct a historical control arm for a clinical trial, and if RWD could be used to complement clinical trial data for this rare pediatric disease.
Overall, the results of this study highlight the limitations of RWD and support the need for clinical trials for this indication.
The U.S. Food and Drug Administration (FDA) initiated this study in the Sentinel System to evaluate utilization of Schedule-II (C-II) stimulant drugs labeled to treat attention-deficit/hyperactivity disorder (ADHD), in light of increasing C-II stimulant use in adults since 2000, which accelerated during the COVID-19 pandemic.1
FDA also sought to gain a clearer understanding of utilization patterns and characteristics of patients starting C-II stimulant therapy, having seen reports of concomitant prescribing of ADHD stimulants with sedative/hypnotic medications or with other psychoactive medications.2
This study assessed baseline characteristics and use patterns of adults starting C-II stimulants from January 2017 to March 2023. Results found the average cumulative treatment duration was approximately six months. A substantial number of patients had observed cumulative treatment duration less than one year. The average daily dose, calculated over all dispensings of each commonly prescribed C-II stimulant, was generally typical of routine clinical practice. Use patterns and baseline diagnoses did not meaningfully differ in the pandemic and pre-pandemic periods. The results of this study provided descriptive information that informed the design of studies assessing risks associated with ADHD stimulant use.
2 Moore TJ, Wirtz PW, Curran JN, Alexander GC. Medical use and combination drug therapy among US adult users of central nervous system stimulants: a cross-sectional analysis. BMJ Open. 2023 Apr 24;13(4):e069668. doi: 10.1136/bmjopen-2022-069668. PMID: 37094897; PMCID: PMC10186433.
The U.S. Food and Drug Administration (FDA) initiated this study to explore the Sentinel System’s ability to assess unmet medical need from drug shortages of certain central nervous system (CNS) stimulants by examining changes over time in the proportion of prescriptions written that were dispensed, using prescribing and dispensing data from several Sentinel Data Partners that were integrated healthcare delivery systems. A secondary analysis examined the proportion of amphetamine/dextroamphetamine dispensings that had a corresponding prescription documented, to assess whether patients who sought care outside of these integrated healthcare systems may have led to the prescribing data missing a substantial amount of the actual prescribing of CNS stimulants. This study was part of FDA’s efforts to understand the extent of the unmet medical need due to shortages in selected prescription stimulants labeled for treating attention-deficit/hyperactivity disorder (ADHD). All prescription stimulants examined were federally controlled substances, schedule II (C-II) of the Controlled Substances Act.
In the secondary analysis of drug prescribing and dispensing data, three-quarters of amphetamine/dextroamphetamine dispensings did not have a corresponding prescription documented in the past 15 days. This result indicated that the available prescribing data missed a majority of all amphetamine/dextroamphetamine prescriptions written for patients in the participating data partners.
Based on this finding, FDA determined that available prescribing data could not support reliable, interpretable estimates of changes over time in the proportion of prescriptions written that were dispensed, to understand unmet medical need associated with shortages of C-II prescription stimulants intended to have been dispensed through outpatient pharmacies. This exploratory study improved FDA’s understanding of the Sentinel Distributed Database’s (SDD) prescribing and dispensing data.
The U.S. Food and Drug Administration (FDA) initiated this study within the Sentinel System to support its response to the coronavirus disease 2019 (COVID-19) pandemic. Early in the pandemic, the FDA recommended that the applicants categorize baseline disease severity in the clinical trial populations using a five-level severity framework. To facilitate regulatory decision-making and post-pandemic evaluations using real-world data, this study aimed to develop operational definitions of COVID-19 severity at diagnosis or at treatment initiation, in ambulatory settings, through code-based algorithms for each severity category based on the FDA’s conceptual guidance. These definitions were designed for use with administrative claims data, a key source of real-world evidence during the pandemic. At the time, no published studies had validated such algorithms for identifying COVID-19 cases or classifying severity within claims data.
The study found that the algorithm demonstrated low to moderate performance in classifying COVID-19 severity in ambulatory settings, with results varying depending on whether severity was assessed at the point of diagnosis or at treatment initiation. While limitations were noted, particularly among untreated populations, the algorithm may be suitable for select study designs, especially those focused on treated patients. This work underscores the Sentinel System’s role in developing new methodological tools and provides a foundation for future surveillance and monitoring leveraging administrative claims data to assess disease severity in emergent public health scenarios.
The U.S. Food and Drug Administration (FDA) initiated this study in the Sentinel System in response to safety concerns raised during the New Drug Application (NDA) review for Sinuva, particularly regarding the potential risks associated with a higher dose of mometasone furoate (MF) and repeated stent use. Sinuva is a mometasone furoate releasing implant used to treat nasal polyps in adults who have previously undergone ethmoid sinus surgery. The objective was to evaluate the incidence of adverse effects among patients receiving a single Sinuva implant compared to those with repeat implants, as well as compared to other MF-eluting implants, such as Propel.
Results indicated that Sinuva usage increased between 2018 and 2021 but declined sharply thereafter, with relatively few patients receiving repeat implants. Among new Sinuva users, rates of glaucoma, ocular hypertension, and cataracts were higher in the Sinuva group compared to Propel. However, the incidence of nasal septal perforation and diminished visual acuity was similar across both groups. These differences appeared to be influenced by age, as Sinuva users tended to be older. When stratified by age, the incidence rates between the two products were comparable. Given the absence of evidence indicating increased risk and the declining use of Sinuva, the FDA concluded that no regulatory action was necessary.
Bictegravir/emtricitabine/tenofovir alafenamide (BIC/FTC/TAF) is currently approved for the treatment of human immunodeficiency virus type 1 (HIV-1). The FDA initiated a study in Sentinel using a self-controlled risk interval analysis and tree-based scan statistics to monitor for new safety concerns among new users of BIC/FTC/TAF. Pre/post and temporal cluster analyses were conducted.
The goal of the study was to monitor non-pregnancy and non-cancer outcomes among new users of BIC/FTC/TAF. After evaluating the statistical alerts generated by this study in the context of other information, the FDA did not identify any new safety concerns and determined that no further evaluation is warranted at this time. Statistical alerts are triaged in consideration of the study design, existing drug knowledge, therapeutic context, treated population, and potential public health impact. Alerts determined to be newly identified safety signals (NISS) follow the FDA’s Center for Drug Evaluation and Research manual of policies and procedures for NISS (MAPP 4121.3). Overall, the study did not identify any new safety concerns for BIC/FTC/TAF in this sample of real-world adult patients.
The U.S. Food and Drug Administration (FDA) initiated this study in the Sentinel System to improve methods for evaluating the safety of medical product use during pregnancy. Accurate identification of pregnancies and estimation of gestational age are critical for assessing prenatal drug exposure. However, data used to identify pregnancies in administrative claims databases generally lack precise indicators of pregnancy start dates, which might lead to wrong conclusions about prenatal drug exposure.
To address these limitations in administrative claims databases, FDA pursued a descriptive analysis to determine whether pregnancy identification could be enhanced by capturing pregnancies without using pregnancy outcome codes (e.g., a code for a live birth) as starting points. This study focused primarily on two analyses. First, certain pregnancy marker codes (including fertility codes, prenatal test codes, and gestational age codes) sometimes are seen in administrative claim records that don’t contain codes for pregnancy outcomes. We evaluated the frequency with which such isolated codes occurred. Second, certain of these pregnancy marker codes are not prioritized by the current Sentinel pregnancy algorithm for determining pregnancy duration and start date. We evaluated the frequency of occurrence of these "lower priority codes" within pregnancies that were identified based on codes describing pregnancy outcomes.
Over 3.97 million females aged 10–54 years with at least one isolated pregnancy marker code in the Merative™ MarketScan® Research Databases were identified between October 2015 and March 2023, including 3.4 million females (86%) without any pregnancy identified. Among the 8.59 million isolated pregnancy marker codes, more than half (52%) occurred in females who remained in the database for over nine months after the code was recorded. The most frequent isolated code was Current Procedural Terminology, Fourth Edition (CPT-4) code 87081 (Group B streptococcus screening), which comprised 4.57 million codes and was non-specific for pregnancy. Additionally, among 2.08 million live birth pregnancy episodes with at least one pregnancy marker code recorded, there were over 25 million lower-priority pregnancy marker codes not used for pregnancy duration estimation, including gestational age codes in 1.57 million episodes (75%) and fertility codes in 44,312 episodes (2%).
The Sentinel study results enhanced FDA’s understanding of the complexities involved in identifying pregnancies and estimating pregnancy start date using administrative claims data. The findings informed discussions on potential methodological improvements to pregnancy identification approaches, but changes to the current algorithm based on these findings will not be implemented at this time. As the FDA pursued this study for the purpose of methods and development, no regulatory action was needed following this study.
Risankizumab-rzaa is a monoclonal antibody and interleukin-23 inhibitor, currently approved for the treatment of adult patients with moderate-to-severe plaque psoriasis, active psoriatic arthritis, moderately to severely active Crohn's disease, and moderately to severely active ulcerative colitis. The FDA initiated two studies in Sentinel using tree-based scan statistics to support safety signal identification for risankizumab-rzaa. A self-controlled risk interval study and an active comparator study using another interleukin-23 inhibitor, guselkumab, as the comparator product were conducted.
The goal of the studies was to monitor non-pregnancy and non-cancer outcomes among new users of risankizumab-rzaa. After evaluating the statistical alerts generated by this study in the context of other information, the FDA did not identify any new safety concerns and determined no further evaluation or action is warranted at this time. Statistical alerts are triaged in consideration of the study design, existing drug knowledge, therapeutic context, treated population, and potential public health impact. Alerts determined to be newly identified safety signals (NISS) follow the FDA’s Center for Drug Evaluation and Research manual of policies and procedures for NISS (MAPP 4121.3). Overall, the studies did not identify any new safety concerns for risankizumab-rzaa in this sample of real-world adult patients.
The U.S. Food and Drug Administration (FDA) conducted this study within the Sentinel System to assess the prevalence of Mayzent (siponimod) exposure among females of childbearing age, both overall and during pregnancy. This evaluation was initiated to investigate the feasibility of continuing two ongoing pregnancy postmarketing requirements (PMRs).
Using data from six Sentinel Distributed Database (SDD) Data Partners with mother-infant linkage capabilities, spanning April 15, 2019, to July 31, 2024, the study identified 772 females of childbearing age who were exposed to siponimod and met the baseline enrollment requirements. This represented 0.16 patients per 10,000 eligible members and accounted for 43.7% of the 1,766 total female siponimod users across all age groups. Among 3,626,656 pregnancy episodes recorded during the study period, fewer than 11 were exposed to siponimod.
These findings supported the FDA’s decision to release PMRs 3591-4 and 3591-5 and instead require a descriptive pregnancy safety study as a new PMR.
Annovera (segesterone acetate and ethinyl estradiol vaginal system) is a new molecular entity approved in 2018 for prevention of pregnancy in women of reproductive potential. During the Phase 3 clinical trial of Annovera, four venous thromboembolism (VTE) cases were observed. This resulted in a crude incidence rate of 24.1 per 10,000 woman-years (95% confidence interval 6.6, 61.7) which was higher than reported for NuvaRing (5.4 per 10,000 woman-years) or other combined oral contraceptives (6.6 per 10,000 woman-years). This prompted a question about whether the risk of VTE is greater for Annovera than for other approved combination hormonal contraceptives (CHCs) in the U.S. The U.S. Food and Drug Administration (FDA) planned this study in the Sentinel System to conduct sequential monitoring to detect any large increase in the risk of VTE or arterial thromboembolism (ATE), particularly as compared to VTE and ATE risk of other approved CHC products. At the time Annovera was approved, Sentinel’s Active Risk Identification and Analysis (ARIA) system was assessed as sufficient for conducting sequential safety monitoring for early detection of a large increase in the risk of non-fatal VTE and ATE.
Although a descriptive analysis was conducted in preparation of the sequential analysis (i.e., calculating incidence and incidence rates of VTE and ATE in new users of CHCs, including NuvaRing), the ARIA study was ultimately not conducted as planned. In parallel with post-marketing surveillance through the ARIA system, the Applicant initially planned an observational study to assess VTE risk and fulfill their postmarket requirement using electronic health record (EHR) data to account for potential confounders such as body mass index (BMI) and smoking. However, lower-than-expected utilization of Annovera limited the feasibility of this approach. As an alternative, the Applicant proposed a study using administrative claims data, supplemented with claims-EHR-linked information on BMI and smoking, and incorporating medical record validation of the outcome algorithm.
Given that the administrative claims data proposed in the revised postmarket required study were comparable to those available in the ARIA system, and claims-EHR-linked data and outcome algorithm validation were incorporated to address limitations inherent in a claims-based study such as that planned in ARIA, the FDA decided to close the sequential analysis of Annovera and VTE/ATE in Sentinel’s ARIA system.
The U.S. Food and Drug Administration (FDA) initiated this study in the Sentinel System to inform the development of a framework to guide the selection of optimal study designs for post-approval pregnancy safety studies. FDA committed to developing this framework under the latest reauthorization of the Prescription Drug User Fee Act (PDUFA VII). The goal of this Sentinel study was to characterize the utilization of a selected group of products during pregnancy and explore product characteristics that may be used to estimate exposure during pregnancy.
A total of 249 products associated with studies in the analysis of post-approval pregnancy safety were identified. Out of the 249 products, all products (n=44) with relatively high exposure (≥2,500) during pregnancies that ended in live births throughout the query period were selected. An additional convenience sample (n=28) of products with relatively low exposure (<2,500) during pregnancies was selected. A total of 72 products were included in the study. Overall, drug utilization in pregnant individuals was low, but the pattern of drug utilization was not solely explained by the number of years on the market. Other factors, such as those related to patient and product characteristics and treatment considerations, should be further explored as potential predictors of drug use during pregnancy.
FDA used results from this study to help identify potential products for the four demonstration projects led by the Center for Drug Evaluation and Research. The demonstration projects are being conducted to meet FDA’s PDUFA VII commitments and will ultimately inform development of the framework to guide optimal study design for post approval pregnancy safety studies.
The U.S. Food and Drug Administration (FDA) initiated this study to assess the feasibility of using mother-infant linked data in the Sentinel System to evaluate the association between in utero exposure to Lipiodol (ethiodized oil) used in hysterosalpingography (HSG) procedures and the development of neonatal thyroid dysfunction, in comparison to in utero exposure to non-oil based iodine agents used for HSG. The Sentinel study was prompted by a study in Japan published in 2015 that reported the frequency of infantile hypothyroidism after exposure to ethiodized oil was higher in their study population (2.4%), compared to 0.7% for congenital hypothyroidism in the Japanese population.
This Sentinel study aimed to monitor Lipiodol use in HSG and infant thyroid dysfunction in the United States. An initial query evaluated the utilization of Lipiodol and non-oil based iodine agents. A very low number of HSG patients with exposure to either Lipiodol or to non-oil agents was observed in relation to the total number of HSG patients. This observation indicated the under-capture of exposure to these contrast agents during a HSG procedure. Therefore, the Active Risk Identification and Analysis (ARIA) System was found to be insufficient to conduct a full study.
The U.S. Food and Drug Administration (FDA) initiated this study in the Sentinel System to assess the feasibility of using Sentinel data to evaluate therapeutic equivalence of brand and generic tacrolimus, potentially resulting in different clinical outcomes between the brand and the generic product. Tacrolimus is an immunosuppressive drug indicated for prophylaxis of organ rejection in patients receiving allogeneic kidney, liver, or heart transplants. This exploratory Sentinel study aimed to assess feasibility of using switching patterns between brand versus individual generic products of tacrolimus oral capsule in transplant patients in the Sentinel Distributed Database (SDD) in order to evaluate therapeutic equivalence.
The first analysis captured liver, kidney, and heart transplant patients in the SDD, and found the majority of patients had less than two years of available follow-up time. The second analysis investigated the continuous use of brand and generic oral tacrolimus following a transplant event. Factors necessary for interpreting switching patterns were only partially captured or uncaptured in claims data (e.g., socioeconomic factors, changes in market share of each product over time, change of pharmacy and availability of generic products at each pharmacy). Given the limited amount of follow-up time available and limited descriptive data for products over time, no additional Sentinel analyses were pursued.
The U.S. Food and Drug Administration (FDA) initiated this study in the Sentinel System to investigate the usage and substitution patterns, as well as clinical outcomes of brand and generic complex drug products. Complex generic products are those that have complex active ingredients, formulations, dosage forms, or routes of administration, or are complex drug-device combinations. Doxorubicin hydrocholoride (HCl) liposome injection – a complex generic drug – is an antineoplastic agent indicated for patients with ovarian cancer whose disease has progressed or recurred after platinum-based chemotherapy; for patients with AIDS-related Kaposi’s sarcoma after failure of prior systemic chemotherapy or intolerance to such therapy; and for patients with multiple myeloma in combination with bortezomib. FDA scientists conducted a feasibility analysis using Sentinel to proactively monitor complex generic drug products in the postmarketing setting. The analysis assessed counts and baseline characteristics of users of brand and generic doxorubicin hydrochloride liposome injection, as well as non-liposomal doxorubicin hydrochloride injection, in the Sentinel Distributed Database (SDD).
Results indicated that most liposomal doxorubicin use in the SDD is identified using Healthcare Common Procedure Coding System (HCPCS) codes as opposed to National Drug Codes (NDCs). Brand products could not be differentiated from generic products using HCPCS codes. No additional Sentinel analyses were pursued because differentiation between brand and generic products, which is critical to this assessment, cannot be achieved with HCPCS codes.
The U.S. Food and Drug Administration (FDA) initiated this study in the Sentinel System to explore whether aggregated electronic health record (EHR) system data could be used to complement administrative claims data in evaluating the safety of non-insulin antidiabetic medications. The study utilized the TriNetX Live™ platform to evaluate the utilization patterns and user demographics for incident new users of specific non-insulin antidiabetic medications, including second-generation sulfonylurea, dipeptidyl peptidase-4 inhibitors, glucagon-like peptide-1 (GLP-1) receptor agonists, metformin, and sodium-glucose co-transporter 2 (SGLT2) inhibitors. Additionally, the study assessed the capture of obesity/overweight diagnoses and body mass index (BMI) for incident new users of GLP-1 receptor agonists, regardless of drug indications.
The results showed utilization patterns for various non-insulin antidiabetic medications. Notably, the results showed that the number of incident new users of GLP-1 receptor agonists and SGLT2 inhibitors increased over time. In the EHR data, race information was available for over 86% of the non-insulin antidiabetic drug users studied. Among all users of GLP-1 receptor agonists in the study, approximately 46% had a recorded Body Mass Index (BMI), while 24% had a documented overweight/obesity diagnosis, both within 183 days prior to their index prescription. These findings indicate that aggregated EHR data captured certain patient demographics, such as race, for a high proportion of patients in our study, suggesting potential for improved capture compared to claims data. However, important characteristics like BMI remained incompletely available in EHR data. Based on the insights gained from this study, future safety evaluations for non-insulin antidiabetic medications could consider integrating EHR data with claims data, particularly for demographics like race. However, for variables like BMI, a careful assessment of EHR data quality and completeness should precede any integration to ensure improved feasibility.
The U.S. Food and Drug Administration (FDA) pursued this study in TriNetX Live™, an aggregated electronic health record (EHR)-based data source, part of the Sentinel System, to learn more about the natural history of coagulopathy in patients with COVID-19. The study aimed to describe individuals hospitalized with COVID-19 who had arterial or venous cardiovascular events (i.e., pulmonary embolism, deep venous thrombosis, stroke, and myocardial infarction) and who were treated with anticoagulants. The study also served to evaluate the feasibility of using real-world EHR data from TriNetX to inform questions related to coagulopathy in COVID-19 patients.
Among ~12,560 hospitalized COVID-19 patients (through June 2020), thrombotic conditions, other COVID-19 severity related conditions, selected medications (days 0-14), and selected laboratory tests (days 0-7) were assessed following their COVID-19 diagnosis or associated hospitalization admission (day 0). Overall, 2.5% of patients had a diagnosis of pulmonary embolism, 2.1% of deep vein thrombosis, 2.4% of ischemic stroke, and 3.9% had a myocardial infarction diagnosis. The most frequent COVID-19 severity related outcome was pneumonia (52.0%). Select medications used in these patients included heparin (20.1%) and low molecular weight heparin (36.1%). Key laboratory data were available for a subset of patients (hemoglobin: 90.8%, d-dimer (FEU): 25.5%, d-dimer (DDU): 17.9%).
The results of this study contributed to the Reagan-Udall Foundation/Friends of Cancer Research Evidence Accelerator discussions with the scientific community, to explore how real-world data (RWD) can quickly address questions on COVID-19 therapeutics. These results also informed an epidemiologic study on the natural history of coagulopathy conducted using data from six Sentinel Data Partners.
FDA initiated this study in the Sentinel System to characterize the utilization of selected drug-device or biologic-device combination products to enhance understanding of factors that may define intended users of the products. Utilization was characterized in the Sentinel Distributed Database (SDD) for individuals 25 years of age or younger with incident use of drug-device or biologic-device combination products for the following drugs or biologics: adalimumab, somatropin, short-acting insulin pens, intermediate-acting insulin pens, long-acting insulin pens, epinephrine, inhaled corticosteroids, nasal corticosteroids, albuterol nebulizer, ophthalmic antibiotic drops, glucagon nasal spray, and leuprolide acetate between January 1, 2018 and February 29, 2020. The drug-device or biologic-device combination products with the highest number of users aged 25 years or younger in the SDD included: albuterol nebulizer (883,533 patients), ophthalmic antibiotic drops (622,693 patients), nasal corticosteroids (355,722 patients), and inhaled corticosteroids (240,114 patients). This analysis captured patient demographic and clinical characteristics, including age, potential indications, health conditions, and intensity of health service utilization, along with product utilization patterns by patient characteristics. Findings informed FDA’s consideration of future studies to better understand use of drug-device or biologic-device combinations, which can support FDA in providing appropriate guidance on these products.
The FDA initiated this study in the Sentinel System to investigate whether in utero exposure to fingolimod, indicated for the treatment of relapsing forms of multiple sclerosis (MS), is associated with an increased risk of cardiac and urinary malformations. This study was prompted by interim reports from the Gilenya Pregnancy Registry, conducted as a postmarketing requirement, which indicated a higher prevalence of congenital malformations, specifically cardiac and urinary, among infants exposed to fingolimod in utero compared to the general U.S. population and infants born to mothers with untreated MS.
The primary objective of this study was to assess the risk of cardiac and urinary congenital malformations in live births from pregnant women exposed to fingolimod during the first trimester of pregnancy. Results indicated an insufficient number of pregnant women exposed to fingolimod during the first trimester or during the 45-day pre-pregnancy period to reach any informative conclusions regarding the risk of congenital malformations.
The results supported FDA’s evaluation of the pregnancy safety signal pertaining to in utero fingolimod exposure.
In 2018, North American and European drug regulatory agencies notified the public about recalls of valsartan and certain other angiotensin II receptor blocker (ARB) products after discovering the presence of potentially carcinogenic nitrosamine impurities, N-nitrosodimethylamine (NDMA) and N-nitrosodiethylamine (NDEA), in the products. There was insufficient data available on how the recall notices impacted valsartan and other ARB utilization in the U.S. and Europe.
The U.S. Food and Drug Administration (FDA) initiated this study to estimate the number of patients affected by these drug recalls and compare the impact of country-specific recalls on valsartan and non-valsartan ARB utilization in the U.S., Canada, the U.K., and Denmark using the FDA’s Sentinel System and international data sources.
Despite the availability of valsartan products without nitrosamine impurities at the time of the recall, results from the U.S., Canada and Denmark revealed a substantial decline in valsartan use following the first recall notices in 2018. Switching from valsartan to the most commonly dispensed ARB in each country appeared to be responsible for the decline. In the U.S., these were most commonly losartan, irbesartan, and olmesartan. Increased switching also was observed around the time of subsequent recalls (after the first notice on valsartan in July 2018) of other affected ARBs, losartan and irbesartan; however, overall usage trends remained unchanged.
Results also demonstrated that despite the widespread use of recalled generic valsartan between 2012 and 2018, individuals’ duration of use was relatively short and likely did not pose an elevated risk of nitrosamine-induced cancer.
These findings provided reassurance that exposure to nitrosamine impurities in valsartan products in the U.S. was limited, and therefore, FDA determined further studies on cancer risk were not necessary.
Velcade (bortezomib) was approved by the U.S. Food and Drug Administration (FDA) in June 2008 for the treatment of multiple myeloma, a cancer of plasma cells. FDA initiated this study in the Sentinel System to explore the feasibility of assessing the rates and timing of the development of health outcomes after initiating bortezomib therapy, focusing on amyotrophic lateral sclerosis (ALS), Alzheimer’s disease, Parkinson’s disease, frontotemporal lobar degeneration (FTLD), amyloidosis, and dementia.
Findings showed that few events were observed among new bortezomib users (N=24,832). ALS had the least (4 events among new users; 2.76 events per 10,000 years at risk); dementia had the most (471 events among new users; 328.75 events per 10,000 years at risk).
The results from this exploratory study informed FDA about the feasibility of identifying the events of interest in the Sentinel Distributed Database.
Following an approximately 2-fold increased risk of lower limb amputations associated with canagliflozin use observed in the CANagliflozin cardioVascular Assessment Study (CANVAS) trial and the CANVAS-Renal trial (a.k.a., CANVAS program), two randomized, placebo-controlled trials, the U.S. Food and Drug Administration (FDA) initiated this study to assess the feasibility of the Sentinel System to investigate the relationship between sodium-glucose co-transporter-2 inhibitor (SGLT2i) use and risk of lower limb amputations in real-world practice across products in the class of SGLT2i.
This feasibility analysis was conducted to assess the overall numbers and person time of new use of SGLT2i (canagliflozin, dapagliflozin, and empagliflozin) and dipeptidyl peptidase-4 inhibitors (DPP4i) (sitagliptin, saxagliptin, linagliptin, and alogliptin), and the overall number of lower limb amputations by age group, sex, and year, during the period between January 1, 2013, and May 15, 2016. Results indicated that there were not enough eligible SGLT2i initiators to provide sufficient statistical power for evaluation of the risk of lower limb amputation either as a class or across individual products in the Sentinel Distributed Database, and the Active Risk Identification and Analysis (ARIA) System was found to be insufficient to conduct an inferential study.
Due to the lack of sufficient statistical power to evaluate risk of lower limb amputation in the Sentinel System, FDA initiated a protocol-based assessment using Medicare data to better characterize the safety concern. Simultaneously clinical trial data became available that led to a 2017 FDA Drug Safety Communication requiring a boxed warning related to this safety concern; however, with more clinical trial data and information from the FDA protocol based assessment using Medicare data and published observational studies that became available, FDA updated the benefit and risk assessment and subsequently removed this Boxed warning in 2020 (2020 FDA Drug Safety Communication).
Cases of cutaneous small-vessel vasculitis (CSVV) associated with direct oral anticoagulants (DOAC) (also known as non-vitamin K oral anticoagulants (NOAC)) were reported in DOAC premarket trials and to the FDA Adverse Events Reporting System (FAERS) post approval. The U.S. Food and Drug Administration (FDA) conducted a study in Sentinel to characterize CSVV cases among patients with atrial fibrillation treated with DOACs. Additional studies in Sentinel explored incidence rates for DOACs, warfarin, and allopurinol as positive control and compared adjusted CSVV risk in new users of DOACs and warfarin. No statistically significant differential CSVV risk was observed among new users of DOACs (dabigatran, rivaroxaban, apixaban) and warfarin. FDA did not take regulatory action at this time, based on available information.
Given the low utilization of edoxaban, the Active Risk Identification and Analysis (ARIA) System was found to be insufficient for studying CSVV after edoxaban exposure.
The U.S. Food and Drug Administration (FDA) initiated this study to evaluate the feasibility of identifying complex generic products and conducting active surveillance of newly approved complex generics within the Sentinel System. Copaxone (glatiramer acetate) was chosen as a test case due to the availability of a product-specific guidance for demonstrating bioequivalence. Both Copaxone and Glatopa (the first generic form of glatiramer), are indicated for the treatment of relapsing forms of multiple sclerosis (MS).
This analysis specifically aimed to assess the utilization of brand and generic forms of glatiramer in MS patients. Results showed 145 patients exposed to Glatopa, 5422 patients exposed to Copaxone 20mg and 4813 patients exposed to Copaxone 40mg from September 21, 2010, to January 31, 2018. Due to low utilization of Glatopa, the FDA determined that Sentinel’s Active Risk Identification and Analysis (ARIA) system was insufficient for conducting an inferential analysis; product utilization could be re-evaluated in the future.
Additionally, this study explored five different algorithms for identifying MS relapse. Algorithms that included diagnoses in outpatient settings captured more MS relapses than algorithms that included diagnoses in inpatient settings alone.
The U.S. Food and Drug Administration’s (FDA) Center for Drug Evaluation and Research (CDER) received an inquiry from another Federal partner requesting information on the impact of the 2023 shortage of injectable methotrexate products on methotrexate utilization, specifically on use among pediatric patients with cancer. FDA conducted this rapid Sentinel study to estimate the utilization of injectable methotrexate among pediatric and adult patients, with and without cancer diagnoses.
The findings from this study showed declines in injectable methotrexate use after shortages were declared, most notably in adult patients without evidence of cancer diagnosis. Among pediatric patients with evidence of cancer diagnosis, injectable methotrexate use appeared relatively steady over time. There was a slight steady decline in use among pediatric patients without evidence of cancer after the shortage was declared. This assessment provided FDA and interested parties insight into the impact of methotrexate injection shortages on patient utilization, specifically for pediatric patients with cancer.
The U.S. Food and Drug Administration (FDA) initiated this study to explore Sentinel’s ability to provide data on the real-world occurrence of thrombotic events in adult patients with COVID-19. FDA intended for these data to inform design decisions, such as sample size estimates, for two National Heart, Lung, and Blood Institute (NHLBI)-funded clinical trials (Accelerating COVID-19 Therapeutic Interventions and Vaccines 4, also known as ACTIV-4) that investigated the efficacy and safety of antithrombotic therapy in preventing thrombotic events in COVID-19 patients in the inpatient and outpatient care settings.
FDA conducted analyses in the outpatient and inpatient care settings using the TriNetX Live™ platform. Among 89,640 adult patients not hospitalized at the time of COVID-19 identification (outpatient analysis) and without evidence of pre-existing risk factors for thrombosis, 0.6% experienced hospitalized thrombotic events or death within 45 days. Only approximately 5% of these COVID-19 patients had relevant laboratory data (C-reactive protein, D-dimer) that was used in clinical trial inclusion criteria.
Among 23,580 hospitalized COVID-19 patients (inpatient analysis), 5.5% experienced a thrombotic event or death within 28 days, a significantly lower frequency of thrombotic events compared with those reported in published literature. Almost half (46.2%) had a D-dimer laboratory result within 3 days, and event rates were similar for patients regardless of D-dimer level (elevated=5.6%, normal=6.4%). The study also aimed to assess the outcome of “free from organ support” but there were substantial challenges in identifying some components of this outcome, particularly oxygen support.
The results of this analysis supported FDA’s public health response to COVID-19. This analysis provided supplemental data to the NHLBI Collaborative Network of Networks for Evaluating COVID-19 and Therapeutic Strategies (CONNECTS) steering and executive committees regarding the thrombotic event rates to support their sample size calculations for the outpatient ACTIV-4 trial.
Tofacitinib, an oral Janus kinase (JAK) inhibitor, was approved in 2012 for the treatment of adult patients with moderately-to-severely active rheumatoid arthritis (RA) who have had an inadequate response or intolerance to methotrexate. The FDA issued a postmarket requirement (PMR) at the time of approval for RA for a controlled clinical trial to evaluate the long-term safety of tofacitinib in patients with moderate-to-severe RA and inadequate response to methotrexate and at least one cardiovascular risk factor to evaluate safety events of interest, including cardiovascular adverse events, opportunistic infections, and malignancy.
Due to safety concerns that arose from this clinical trial, FDA conducted an analysis in the Sentinel System to better understand the characteristics of users of tofacitinib compared to other disease-modifying anti-rheumatic drugs (DMARD) - conventional DMARDs, JAK inhibitors, tumor necrosis factor (TNF) inhibitor or other biologics users-, with a focus on RA severity and utilization patterns. Results indicated RA severity could not be reliably identified from the Sentinel Distributed Database’s administrative claims data alone. Given that identifying RA severity was critical to this assessment, no additional Sentinel analyses were pursued.
After a comprehensive review of the available information, the FDA added a Boxed Warning to tofacitinib’s label in July 2019, indicating that “rheumatoid arthritis patients with at least one cardiovascular (CV) risk factor had a higher rate of all-cause mortality and thrombosis with XELJANZ 10 mg twice daily vs. 5 mg twice daily or TNF blockers.”
The U.S. Food and Drug Administration (FDA) initiated this study in the Sentinel System to examine the potential exposure of pregnant women to ibrexafungerp. Ibrexafungerp, a first-in-class agent of a new antifungal class (triterpenoid), is approved for treatment of vulvovaginal candidiasis and for reduction in the incidence of recurrent vulvovaginal candidiasis in adult and post-menarchal pediatric females. Ibrexafungerp is contraindicated during pregnancy due to potential fetal harm, based on findings from animal reproductive studies. However, most ibrexafungerp users are expected to be women of reproductive age. This Sentinel study aimed to actively monitor postmarket exposure to ibrexafungerp during pregnancy.
Between June 1, 2021, and August 31, 2023, results indicated that out of 385,507 eligible pregnancy episodes, three pregnancies were potentially exposed to ibrexafungerp during the first trimester when assuming a 7-day supply. When a 30-day supply was assumed, seven pregnancies were potentially exposed during the first trimester. However, these exposures cannot be definitively confirmed due to known limitations of the rapid Sentinel database. Some database limitations include not having confirmation of whether the patient consumed the drug and the algorithms used may have misclassified pregnancy start and duration. This study supported the enhanced monitoring of ibrexafungerp utilization to identify any potential exposure to ibrexafungerp during pregnancy.
Guselkumab is a monoclonal antibody and interleukin-23 inhibitor currently approved for the treatment of adult patients with moderate-to-severe plaque psoriasis, active psoriatic arthritis, or moderately to severely active ulcerative colitis. The FDA initiated two studies in Sentinel using tree-based scan statistics to support safety signal identification for guselkumab. A self-controlled risk interval study and an active comparator study using another interleukin-23 inhibitor, risankizumab-rzaa, as the comparator product were conducted.
The goal of the studies was to monitor non-pregnancy and non-cancer outcomes among new users of guselkumab. The FDA determined the statistical alerts generated by these analyses did not warrant further evaluation or action. Overall, the studies did not identify any new safety concerns for guselkumab in this sample of real-world adult patients.
The FDA initiated a study in the Sentinel System to estimate the prevalence of prurigo nodularis in order to assess the feasibility of conducting studies on products indicated for prurigo nodularis in pediatric patients. There is limited knowledge about age-specific incidence and prevalence of prurigo nodularis, particularly in pediatric age groups.
Sentinel System results described prurigo nodularis as a rare condition among children that is diagnosed much less frequently in children than adults. Prurigo nodularis prevalence (in 2022) increased progressively by age, 0.02-0.17 per 1,000 in pediatric age groups (age <18 years) to 3.12 per 1,000 in older adults (age ≥65 years). These results suggest that the feasibility of studies of new prurigo nodularis treatments in pediatric patients may be low as prurigo nodularis in children is rare. These findings informed the FDA’s decisions on sponsor-required studies in pediatric patients.
The U.S. Food and Drug Administration (FDA) pursued this study to characterize the TriNetX Live™ platform data within the Sentinel System, as these data may be used for future Sentinel studies. FDA sought to better understand attributes of the available data, including the extent to which Health Care Organization (HCO) data had date shifting (a part of data sources’ privacy preserving measures) and whether date shifting may impact future study approaches.
Results provided demographic and geographic patterns of data stratified by whether the data sources underwent date shifting. As the FDA pursued this study for the purpose of methods and development, no regulatory action was needed as a result of this study.
Dupilumab is a monoclonal antibody used for management of atopic dermatitis, asthma, chronic rhinosinusitis with nasal polyps, eosinophilic esophagitis, prurigo nodularis, and Chronic Obstructive Pulmonary Disease. The FDA initiated a study in Sentinel using a self-controlled risk interval analysis and tree-based scan statistics to monitor for new safety concerns among new users of dupilumab. A pre/post and a temporal cluster analysis were conducted.
After evaluating the statistical alerts generated by this study in the context of other information, the FDA determined no further evaluation is warranted at this time. Statistical alerts are triaged in consideration of the study design, existing drug knowledge, therapeutic context, treated population, and potential public health impact. Alerts determined to be newly identified safety signals (NISS) follow the FDA’s Center for Drug Evaluation and Research manual of policies and procedures for NISS (MAPP 4121.3).
Observational studies assessing drug safety are usually conducted among incident new users of medications. However, recent studies have considered using cohorts of prevalent new users to evaluate the safety of antidiabetic drugs such as sodium-glucose co-transporter-2 inhibitors (SGLT2i) to increase sample size and expand the evaluation of the safety of antidiabetic drugs to all users. The U.S. Food and Drug Administration (FDA) initiated this study in the Sentinel System to characterize factors related to the assignment of antidiabetic treatments among patients with type 2 diabetes mellitus with and without a history of non-metformin antidiabetic medication use to understand the impact of using prevalent new users in safety evaluations of SGLT2i.
Results found that factors related to the choice of SGLT2i over dipeptidyl peptidase-4 inhibitors (DPP-4i) differ substantially among naïve, incident, and prevalent new users. SGLT2i initiators were more likely than the DPP4i users to have baseline non-metformin antidiabetic medication use in both the prevalent new users and incident new users except for sulfonylureas. Observational studies that include prevalent new users require proper design and analytic approaches to address imbalances among these populations in order to avoid bias. This study highlights that methodology to properly account for the possible imbalances within the study population should be carefully considered when studies that assess the safety of SGLT2i include prevalent antidiabetic medication new users to increase the study sample size. This study supported the FDA in interpreting studies on the safety of SGLT2i which included prevalent new users.
The FDA conducted analyses in the Sentinel System to describe medication use and evaluate the risk of adverse infant and maternal outcomes among pregnant patients with coronavirus disease 2019 (COVID-19). These analyses supported an international collaboration funded by the European Medicines Agency (EMA) to study COVID-19 and pregnancy, known as “COVID-19 infectiOn aNd medicineS In pregnancy” (CONSIGN)1
. The CONSIGN study, which drafted a protocol to examine the natural history of COVID-19 disease in pregnant patients, was implemented across data sources in eight European countries. This Sentinel study implemented the following three CONSIGN protocol aims:
To estimate the prevalence of medicines used and compare this among pregnant patients with COVID‐19, pregnant patients without COVID‐19, and nonpregnant patients with COVID‐19.
To describe severity and clinical outcomes of COVID‐19 disease in pregnant patients with COVID‐19, according to treatments received during pregnancy, and compare these data with those of nonpregnant patients of reproductive age with COVID‐19.
To assess and compare the rates of adverse maternal and neonatal outcomes in cohorts of pregnant patients with COVID-19 diagnosis in the first, second, or third trimester during pregnancy and pregnant patients without COVID-19.
Sentinel results indicated that medications used for potential COVID-19 treatment varied over time. The analyses did not find an increased risk of adverse maternal or infant outcomes in pregnancies with COVID-19 compared to those without COVID-19. However, among less than 5% of pregnancies with severe COVID-19 in the third trimester, a higher proportion of low birth weight was observed. Additionally, higher rates of preterm birth and caesarean section were noted among pregnancies with severe COVID-19. These findings may be challenging to interpret due to temporal changes in COVID-19 severity, treatment, and prevention advancements. The results of the Sentinel analyses contributed to a meta-analysis conducted across ten healthcare data sources in Europe, the United States, and Canada.
The objective of this study was to explore the FDA Sentinel System’s ability to conduct real-world evidence evaluations for the pediatric population, with a focus on identifying and comparing clinically-defined and claims-based pediatric hypertensive patients within the Sentinel System.
Given concerns about the potential under-capture of pediatric hypertension in claims-based data observed in the initial analysis, the study further explored the use of blood pressure measures for identifying hypertension in the pediatric population. The follow-up analysis compared and examined overlap between the two methods for identification of pediatric hypertensive patients (claims-based and clinical electronic health record [EHR]-based) over a 36-month period in the Sentinel System. Findings indicated poor concurrence between claims and clinical definitions of hypertension. Pediatric hypertension is under-captured in claims-based data sources but can serve as a marker for greater disease severity.
This study provided FDA with insights into the differences in how pediatric hypertension is captured across different real-world-data (RWD) sources, which could support the planning of future pediatric hypertension studies.
In the United Kingdom Randomized Evaluation of COVID-19 Therapy (RECOVERY) trial results publicized in June 2020, dexamethasone appeared to reduce mortality in hospitalized patients with COVID-19 who received supplemental oxygen or mechanical ventilation. However, a potential signal of harm of dexamethasone use was seen in hospitalized patients with COVID-19 who did not receive respiratory support. To gain insight into how the trial results may affect inpatient corticosteroid use patterns in the U.S., the Food and Drug Administration (FDA) conducted a rapid study to assess the utilization of systemic dexamethasone and other corticosteroids among hospitalized patients with and without a COVID-19 diagnosis in the TriNetX USA Network Database, one of the FDA’s Sentinel Electronic Health Record (EHR) data sources.
Results showed that among hospitalized patients with COVID-19, the weekly proportion administered corticosteroids, dexamethasone in particular, increased substantially from the week beginning June 8, 2020 to the week beginning July 13, 2020; administration of corticosteroids remained stable among hospitalized patients without COVID-19. This analysis provided data to the FDA that may have helped inform strategies for allocating existing drug supplies to promote patient care during the COVID-19 pandemic.
As part of the activities of the FDA’s Cannabis Product Committee, the Office of Surveillance and Epidemiology, Center for Drug Evaluation and Research, conducted an integrated review to evaluate and compile an updated catalog of available data sources that complement FDA’s routine surveillance systems for monitoring non-FDA-approved cannabinoid products (CP) use and associated adverse events. FDA explored the capabilities of the Sentinel System’s electronic health record (EHR) data, specifically in TriNetX and PCORnet, to conduct active safety surveillance of CP, including cannabidiol (CBD). Results from TriNetX analyses suggest the ability to extract and identify non-FDA-approved CP exposures from clinical notes. There are opportunities to enhance the performance of pertinent natural language processing (NLP)-based algorithms to improve the identification of CP in these notes. Results from PCORnet suggest that identified CBD exposures correspond to approved CBD (Epidiolex). Future explorations on FDA-approved CBD in PCORnet should consider including exposures with and without brand names to capture exposures to FDA-approved CBD that might be overlooked. Further development and validation of NLP-based algorithms is needed before FDA can consider use of data from EHR clinical notes for surveillance of non-FDA-approved CP.
The FDA initiated this study to understand the differences in how key covariates are captured in electronic health records (EHR) versus claims data. This study assessed the identification of key covariates, such as body mass index (BMI) and smoking status, among users of sodium-glucose co-transporter 2 (SGLT2) inhibitors and sitagliptin. This examination was conducted using EHR data from the National Patient-Centered Clinical Research Network (PCORnet) as well as claims and Integrated Delivery System (IDS) data in the Sentinel Distributed Database, from March 1, 2013, through June 30, 2018.
EHR data identified more patients with documented records of BMI and smoking status than claims data. An IDS analysis of new SGLT-2 inhibitor users observed a good concordance (77%) between claims and EHR data for current tobacco use. A more sensitive definition of obesity in claims data increased concordance (44%) with EHR data, compared to a narrower, BMI-specific definition (14%).
This methods project characterized missingness and clinical measurement concordance between EHR and claims data for key covariates, which could inform quantitative bias analysis or imputation approaches for FDA’s drug safety assessments.
The U.S. Food and Drug Administration (FDA) initiated this study in the Sentinel System to estimate the prevalence of 59 pre-defined health conditions (using the Centers for Medicare and Medicaid Services' Chronic Conditions Data Warehouse criteria) among individuals with childbearing potential in the Sentinel Distributed Data. The prevalence of the pre-defined health conditions are an essential element to develop a tool to estimate potential medical product utilization upon approval among individuals with childbearing potential. Potential for product utilization upon approval is an important element to consider when determining the types of postmarket pregnancy safety studies. This work commenced to support the Prescription Drug Use Fee Act (PDUFA) VII Pregnancy Demonstration Projects.
The prevalence of the selected health conditions among individual with childbearing potential varied considerably. Anxiety disorder, depression, obesity, and hypertension were the conditions with the highest prevalence. The prevalence of the selected health conditions varied by age group and year.
The findings from this study will be considered to develop an algorithm to estimate potential medical product utilization upon approval among individuals with childbearing potential. This will support identification of the most suitable study approaches to assess medical product safety among pregnant patients.
The U.S. Food and Drug Administration (FDA) initiated this study in the Sentinel System to assess whether there is a signal of carcinogenicity for bumetanide due to nitrosamine-related impurity using epidemiological data. N-Nitroso bumetanide can form as a degradation impurity in some formulations of bumetanide during shelf-life. Due to its structural similarity to other nitrosamine genotoxic impurities, it is potentially genotoxic.
This study explored whether bumetanide is associated with an increased risk of cancer compared to other loop diuretics and the background population.
Results from Sentinel analyses did not demonstrate an increased risk of four biologically plausible cancer outcomes (liver, colorectal, bladder and esophageal cancers) among bumetanide users compared to users of other loop diuretics. Similarly, a dose-response signal was not suggested by the findings.
Results from this study were shared with FDA’s nitrosamine task force as part of their efforts to address nitrosamine-related impurities in human drug products.
Labeling for Human Epidermal Growth Factor Receptor 2 (HER2) antagonist products includes a boxed warning describing embryo-fetal toxicity associated with oligohydramnios. The FDA initiated this study to determine the real-world exposure rates to HER2 antagonists in pregnancy to better understand the scope of risk for the labeled adverse outcomes. The Merative™ MarketScan® Research Database was utilized to assess how many people have been exposed to HER2 antagonists in pregnancy. Results identified 24 pregnancy episodes with HER2 antagonist exposure among 1,737,836 live births in the Merative™ MarketScan® Database from January 1, 2012, to September 30, 2019. The proportion of exposed pregnancies does not surpass those reported in other post-marketing data and previous literature.
This analysis was also used to inform the feasibility of assessing the risk of oligohydramnios after HER2 antagonist exposure during pregnancy using the Sentinel System. Given the low exposure rate, the Active Risk Identification and Analysis (ARIA) System was found to be insufficient for studying oligohydramnios after HER2 use during pregnancy.
The U.S. Food and Drug Administration (FDA) initiated this study in the Sentinel System to support the Office of Surveillance and Epidemiology’s active safety monitoring plan for therapeutics granted Emergency Use Authorization (EUA) status for the treatment of coronavirus disease 2019 (COVID-19). EUA authority allows FDA to strengthen the nation’s public health protections by authorizing unapproved medical products or unapproved uses of approved medical products during public health emergencies, such as the COVID-19 pandemic. The COVID-19 EUA safety monitoring plan aims to provide a mechanism for capturing serious adverse events (known, suspected, or previously unknown) during the EUA period. The goal of the monitoring plan is signal identification and characterization.
FDA launched a series of analyses over the COVID-19 pandemic to periodically assess whether the use of several therapeutics for COVID-19 treatment under EUA followed the authorized use of these products, as well as describe patient outcomes, including anaphylaxis and hospitalization rates, after receiving outpatient COVID-19 treatments under EUA. Additionally, FDA sought to estimate the magnitude of the “at-risk” population who used drugs that interact with Paxlovid (nirmatrelvir and ritonavir) to inform product labeling.
In total, FDA assessed the following COVID-19 EUA therapeutics: bamlanivimab, bamlanivimab/etesevimab, baricitinib, bebtelovimab, casirivimab/imdevimab, molnupiravir, nirmatrelvir and ritonavir, remdesivir, and sotrovimab. The periodic EUA monitoring analysis suggested that the COVID-19 EUA treatment use followed the Fact Sheet directives and did not identify new safety signals. The drug interaction assessment indicated that more than 50% of Paxlovid-eligible patients may have been on drugs that could interact with Paxlovid. However, most of the commonly used drugs that are involved in these drug-drug interactions with Paxlovid do not seem to prevent patients from taking Paxlovid, as they can be temporary withheld or adjusted to lower dose. Prescribers should be aware of the risk of drug interaction and refer to the Paxlovid label and other appropriate resources for comprehensive information on the drug interaction risk management.
Findings from the drug interaction assessment were included in the FDA’s New Drug Application review of Paxlovid, which emphasizes adverse reactions as a key component of the safety review. After reviewing the totality of the information available, FDA added a boxed warning to the Paxlovid label to underscore the drug interaction risk.
Entyvio (vedolizumab) is an integrin receptor antagonist indicated for the treatment of adult patients with moderately to severely active ulcerative colitis and Crohn’s disease, the two main disorders that comprise inflammatory bowel disease (IBD). Based on two previous Periodic Safety Reports, the European Medicines Agency (EMA) evaluated cases of interstitial lung disease (ILD) in vedolizumab users and concluded that the contribution of vedolizumab could not be excluded. There are also limited available data on the background incidence rates of ILD in patients with IBD, as well as the incidence rates of ILD in IBD patients treated with vedolizumab or other biologics. Due to this, FDA issued a Newly Identified Safety Signal (NISS) in January 2023 to facilitate evaluation and management of this potential risk for ILD. Tysabri (natalizumab), another integrin receptor antagonist indicated for treatment of multiple sclerosis and Crohn’s disease, was also evaluated for ILD as part of this NISS. The FDA pursued studies in the Sentinel System to better understand the incidence of ILD in IBD patients treated with vedolizumab, natalizumab, or other advanced therapies. Specifically, Sentinel System studies estimated the incidence rate of ILD among patients with IBD overall and separately among patients treated with vedolizumab, natalizumab, or other advanced therapies.
Sentinel System results did not indicate an increased incidence rate of ILD in patients treated with vedolizumab or natalizumab for IBD. The incidence rates of ILD for patients with IBD treated with integrin receptor antagonists approximates the ILD incidence rates observed separately in IBD patients who initiated and actively used advanced therapies other than an integrin receptor antagonist or patients with IBD with a history of treatment with advanced therapies other than an integrin receptor antagonist.
As part of the NISS evaluation, FDA used findings from the Sentinel analysis to support contextualizing the identified safety signal and provide additional data on the burden of disease. Based on the entire body of evidence evaluated as part of the NISS, including 18 postmarketing cases of non-infectious ILD with vedolizumab or natalizumab in the FDA Adverse Event Reporting System (FAERS) database and 21 cases of ILD in vedolizumab-treated patients published in the medical literature, FDA approved a safety-related labeling supplement request for vedolizumab on April 18, 2024. This safety-related labeling update incorporated the terms, “interstitial lung disease, pneumonitis” into the Postmarketing Experience subsection of the Prescribing Information.
The U.S. Food and Drug Administration (FDA) used the Sentinel System to assess incidence and prevalence of medical codes associated with local anesthetic toxicity. This study specifically assessed International Classification of Diseases, Ninth Revision, Clinical Modification (ICD-9-CM) codes.
The results included counts and prevalence of three diagnostic ICD-9-CM codes for local anesthetic toxicity stratified by year, sex, and age group. Prevalence of local anesthetic toxicity ranged from 0 to 1.02 per 100,000 enrollees, varying by year, sex, or age group.
This exploratory study provided information to FDA on the incidence and prevalence of local anesthetic toxicity medical codes in the Sentinel Distributed Database, to better aid decisions on the use of Sentinel data for capturing local anesthetic toxicity.
The Centers for Disease Control and Prevention (CDC) collaborated with the Food and Drug Administration (FDA) to explore whether the Sentinel Distributed Database (SDD) could enhance CDC’s and FDA’s understanding of the epidemiology of latent tuberculosis infection (LTBI). This CDC-FDA collaborative study assessed LTBI testing, clinical management, and treatment duration in individuals treated for LTBI.
Among 113,338 patients who filled prescriptions for drugs indicated for LTBI, INH-only was the most commonly prescribed LTBI treatment with 80% (90,377) receiving isoniazid (INH) only, 19% (21,235) rifampin (RIF) only, and 2% (1,726) INH + rifapentine (RPT). Overall, 88% of patients had at least one element of diagnostic evaluation documented before starting treatment. Most persons who filled a prescription for LTBI treatment did not have evidence of completing the recommended treatment durations. The data further support preferential use of shorter-course regimens such as INH + RPT.
Using Sentinel data, this study was able to describe the common LTBI treatment regimens and provide insight into prescribing behavior. The study also showed the treatment duration for each regimen, highlighting the need for additional interventions to support patients’ completion of/adherence to LTBI therapy. Additionally, the study identified varying rates of diagnostic testing among LTBI patients by treatment regimen, suggesting a potential lack of provider familiarity with recommendations.
Due to limitations in this study common to claims-based data sources, such as lack of information on tuberculosis related test results, incomplete race and ethnicity data, and restriction to individuals with health insurance, FDA’s Sentinel System may not be suitable for general population-level LTBI surveillance in the United States. Nevertheless, the FDA Sentinel System can serve as a complementary source of data to augment surveillance of adverse events related to LTBI treatment regimens, describe specific groups of individuals with LTBI, and provide insight into prescribing patterns for LTBI.
In November 2020, the U.S. Food and Drug Administration (FDA) issued an emergency use authorization (EUA) for baricitinib to be used in combination with remdesivir to treat COVID-19 in hospitalized patients on respiratory support (i.e., supplemental oxygen, non-invasive or invasive mechanical ventilation, or extracorporeal membrane oxygenation). In June 2021, the FDA issued an EUA for tocilizumab to treat COVID-19 in hospitalized patients receiving systemic corticosteroids and on respiratory support. To understand the scope of utilization and characteristics of users of baricitinib and tocilizumab in the context of COVID-19, FDA conducted a descriptive analysis among patients with inpatient baricitinib or tocilizumab use in the Sentinel System. Both products were commercially available prior to FDA issuance of the EUAs, and the study period for this analysis (April 1, 2020, to March 31, 2021) included time prior to FDA issuance of these EUAs. Specific characteristics assessed included evidence of COVID-19, use of respiratory support, concomitant drug use, and medical conditions.
Results indicated that most patients who received inpatient baricitinib/remdesivir therapy had evidence of COVID-19. However, about half of patients who received inpatient baricitinib therapy did not appear to have concurrent remdesivir use, and few patients with inpatient baricitinib therapy appeared to have documented respiratory support at drug therapy initiation. Results also indicated that most patients with inpatient tocilizumab therapy had evidence of COVID-19 and a majority had concurrent systemic corticosteroid therapy. Additionally, around 25% of patients with tocilizumab therapy had documented mechanical ventilation at drug therapy initiation. This analysis informed the FDA’s active safety monitoring efforts for products authorized or used for the treatment of COVID-19. FDA approved these products for the treatment of adults with COVID-19 in May 2022 (baricitinib) and December 2022 (tocilizumab), and EUAs remain in place for use in pediatric patients as of August 2023.
The FDA pursued this study to demonstrate how Sentinel can be utilized to answer regulatory questions regarding drugs with elements to assure safe use (ETASU) by exploring the utilization patterns of drugs with approved Risk Evaluation and Mitigation Strategies (REMS) that employ ETASU. To ensure that the benefits of a drug or biological product outweigh its risks, the FDA can require a REMS with an ETASU to mitigate a specific and serious risk listed in the labeling of the drug. This study provided summary tables of total counts, incidence, and prevalence of dispensings and procedure codes of drugs with approved REMS that employ ETASU among enrolled members in the Sentinel Distributed Database (SDD). This study provided baseline information on utilization patterns of specific drugs with REMS with ETASU which could support the planning of future studies on these products.
Low-dose oral methotrexate is associated with wrong frequency dosing errors, when taken once daily instead of the intended once weekly schedule. The Institute for Safe Medication Practices (ISMP) has classified methotrexate (oral, nononcologic use) as a high alert medication that can cause fatal and serious adverse events when mistakenly taken daily. However, the incidence of wrong frequency dosing errors with methotrexate is unknown. A chart confirmed analysis was conducted in one Sentinel Data Partner (Kaiser Permanente Northern California) and found the incidence of low-dose oral methotrexate wrong frequency dosing errors to be 0.4%. FDA used these findings to revise the methotrexate labeling in 2019, which included adding a new Warnings and Precautions section on the risk of improper dosing, removing an option for doses given every 12 hours for 3 days each week, and recommending that patients and caregivers be instructed to take methotrexate as directed as dosing errors have led to fatal toxicity. In July 2020, ISMP highlighted the labeling revision in their Medication Safety Alert! newsletter.
In July 2015, FDA released a drug safety communication regarding prescribing or dispensing errors due to brand name confusion with the antidepressant Brintellix (vortioxetine) and the antiplatelet Brilinta (ticagrelor). In May 2016, FDA approved a brand name change for the antidepressant Brintellix to Trintellix.
This study was intended to explore Sentinel’s ability to detect potential drug name confusion errors using Brintellix-Brilinta as a feasibility use case. We developed a claims-based algorithm for identifying potential drug name confusion errors. In combination with claims profile review, we identified likely errors in both Brintellix and Brilinta users. This study demonstrated that a claims‐based algorithm combined with manual review of claims profiles could be used to identify potential drug name confusion errors.
The U.S. Food and Drug Administration (FDA) initiated this study in the Sentinel System to describe the natural history of enterovirus sepsis among infants and neonates in the United States. Enterovirus (EV) infection in neonates can manifest as asymptomatic or mild febrile illness, or as a severe multiorgan system disease. The epidemiology of neonatal enteroviral sepsis (NES) in the United States is not well characterized, and likely varies by geography, enterovirus serotype, population, season, and testing practices. To further understand the epidemiology of NES, this study examined the prevalence of NES among hospitalized infants and described the associated mortality rate.
Results indicated that NES was rare with a prevalence of 0.83 per 100,000 inpatient stays. Hospital stay was longer among cases with NES and at least one organ dysfunction diagnosis. No deaths were identified. This was the first study to examine NES prevalence using inpatient data from the Sentinel System. Due to small sample size and lack of microbiology/virology data, its utility in informing future clinical trial designs was limited. No regulatory action was taken based on these data.
Hydroxyurea was approved in 1967 for treating certain types of cancer. In 1998, the drug was approved to reduce the frequency of painful crises and to reduce the need for blood transfusions in adult patients with sickle cell disease (SCD) with recurrent moderate to severe painful crises. Hydroxyurea was approved for pediatric use by FDA in 2017 to reduce the frequency of painful crises and to reduce the need for blood transfusions in pediatric patients, two years of age and older, with sickle cell anemia with recurrent moderate to severe painful crises. Since the pediatric approval, little has been published about hydroxyurea utilization patterns in pediatric populations. FDA conducted three analyses in the Sentinel Distributed Database, an analysis using data from 2000 to 2015 and two analyses using data from 2000 to 2020. In these analyses, FDA assessed the utilization of hydroxyurea by age group and indication and evaluated the number of hospitalizations/emergency department (ED) visits in patients with SCD for two time frames: (1) any observed time up until a patient’s first hydroxyurea dispensing, and (2) among patients with hydroxyurea use, observed time following their first hydroxyurea dispensing.
The two analyses which used data from 2000 to 2020 entailed slightly different cohort entry requirements. In the first analysis, Sentinel results identified 113,036 patients of any age with SCD who were active in the data source sometime between January 2000 and September 2020. Of these patients, 16,483 patients (15%) were ever dispensed hydroxyurea. Among the 45,472 African American patients with SCD, 10,339 patients (23%) had hydroxyurea use, compared to 13% of patients with an unspecified race and 2% of Caucasian patients. A total of 96,297 adult patients 18 years old or older with SCD were identified, 13,740 (14%) of whom were dispensed hydroxyurea. Over three-quarters of adult patients had at least one hospitalization or ED visit (for any medical condition). The analysis showed 29.4 hospitalizations or ED visits per 10 person-years among adult patients with SCD while not dispensed hydroxyurea, and 79.4 hospitalizations or ED visits per 10 person-years among adult patients with SCD after being dispensed hydroxyurea. Of note, disease severity was not accounted for in this analysis.
This analysis also identified 16,739 pediatric patients aged 17 years old and younger with SCD, of whom 2,743 patients (16%) were ever dispensed hydroxyurea. Among all pediatric patients with SCD, 11,457 patients (68%) had at least one hospitalization or ED visit (for any medical condition, 15.8 events per 10 person-years). While not dispensed hydroxyurea, pediatric patients with SCD had 12.9 hospitalizations or ED visits per 10 person-years, compared to 30.5 hospitalizations or ED visits per 10 person-years after being dispensed hydroxyurea (not accounting for disease severity).
The second analysis identified 14,294 patients of all ages with SCD who had health plan enrollment during the 183 days prior to their first observed hydroxyurea dispensing, of whom 5,872 patients (41%) received at least one year of therapy, cumulatively. Approximately half of the pediatric patients 17 years old or younger who received hydroxyurea received at least one year of therapy, cumulatively, compared to 39% of adult patients 18 years old or older having received at least one year of therapy.
These analyses informed FDA about real-world utilization patterns for hydroxyurea among pediatric and adult patients with SCD from 2000 to 2020.
In an earlier FDA study of intravenous (IV) iron products, differences in the market share of specific IV iron products were detected between patient-level administrative claims data from Sentinel and national manufacturer sales data. To examine whether the differences were due to challenges in estimating utilization for products with a non-oral route of administration, such as those involving injections, FDA assessed the agreement between data sources and utilization metrics by evaluating market share for selected drug classes. Six classes of drugs were selected, three orally administered drugs and three drugs with more complicated routes of administration. The study included data from January 1, 2011 to December 31, 2015. Agreement between patient-level data and national manufacturer sales data varied across different metrics, in part driven by product characteristics. The study helped FDA staff to understand the importance of comparing different utilization metrics when assessing medication use across multiple data sources and selecting the utilization metric that best aligns with how products are used and what is intended to be measured.
In October 2019, FDA pursued a study in the Sentinel System to describe utilization patterns for spironolactone in patients with heart failure (HF) with preserved ejection fraction (HFpEF). The study was initiated as the FDA Center for Drug Evaluation and Research (CDER) Division of Cardiology and Nephrology was considering data from a clinical trial sponsored by the National Institutes of Health (NIH) that found a significantly lower incidence of heart failure (HF) hospitalization among HFpEF patients treated with spironolactone compared to placebo. To characterize the real-world utilization of spironolactone in patients with HFpEF, and in patients with HF with reduced ejection fraction (HFrEF), FDA conducted a study in the Sentinel System. The study identified 2,009,529 patients with HFrEF and 9,257,514 HFpEF patients. The proportion of patients initiating spironolactone was 20.7% after HFrEF diagnosis versus 7.6% after HFpEF diagnosis. The median time (days) to initiation of spironolactone after HFrEF diagnosis was 90 (IQR: 19–385) and after HFpEF diagnosis was 286 (IQR: 57–851). The median duration (days) of first treatment episode in HFrEF patients was 120 (IQR: 44–321) and for HFpEF patients was 114 (IQR: 32 – 301). The median dose was similar (25mg/day) for both HF cohorts. This study characterized the use of spironolactone in a large database with demographically and geographically diverse patients. While there was lower initiation of spironolactone following HFpEF compared to HFrEF diagnosis, the dosing and duration of the first continuous spironolactone episode were similar. As a result of this study, no regulatory action was taken.
This study was conducted to assist the U.S. Food and Drug Administration (FDA) in reviewing several requests for orphan drug designations by estimating the number of individuals affected by several glomerular diseases in the United States.
Data from this study suggested glomerular diseases seemed to be rare in the United States. Based on the finding of this study in the FDA’s Sentinel System and Census data projections, numbers of individuals with focal segmental glomerulosclerosis (FSGS), IgA nephropathy (IgAN), membranous nephropathy (MN), minimal change disease (MCD), membranoproliferative glomerulonephritis (MPGN), crescentic and necrotizing glomerulonephritis (CNGN), lupus nephritis (LN), and nephrotic syndrome (NS) in the United States were all below the 200,000 person threshold for orphan drug designation, for each year from 2016 to 2019.
Nevertheless, because algorithms used to estimate different glomerular diseases were not validated, estimates are subject to potential case misclassification, which may underestimate or overestimate the true disease prevalence.
In the development program, vericiguat showed embryo-fetal toxicities in animal studies which may be associated with the soluble guanylate cyclase stimulator class. The FDA review team sought information to inform whether a Risk Evaluation and Mitigation Strategies (REMS) program should be required for vericiguat. A retrospective cohort study was conducted in the Sentinel System to assess prevalence of heart failure (HF) in women of childbearing age and the number of live birth pregnancies among these women. A second analysis described characteristics and outcomes of pregnancies in women with HF, and compared them to age-matched pregnant women without HF.
From January 2010 to February 2020, 144,162 women with HF were identified (prevalence, 0.5%) among 29.5 million women of childbearing age in the Sentinel System. Within this HF cohort, there were 813 women with 822 pregnancies ending in live birth deliveries (5.5 deliveries per 1,000 women with HF). Applying the prevalence of HF to the 2019 Census estimates, we projected there were 310,613 women of childbearing with HF in the U.S. in 2019 and among these women, there were an estimated 808 pregnancies ending in live birth deliveries.
In the second analysis, 489 live birth deliveries were identified (mean maternal age, 32.4 years) in 487 women with HF. These women had more comorbidities and used more health services than pregnant women without HF but were healthier than age-matched non-pregnant women with HF. Beta-blockers (21.5%), diuretics (15.3%), and ACE inhibitors (10.2%) were the commonly used HF medications during the pre-pregnancy period. Utilization of beta-blockers remained unchanged throughout and after pregnancy. Use of ACE inhibitors dropped to 5.3% in the first trimester, to less than 1% in the second and third trimesters but increased to 8.0% after pregnancy. Use of other HF medications, such as aldosterone antagonists, angiotensin receptor blockers, and ivabradine, were rare.
HF is rare among women of childbearing age and pregnancies only occurred in a small number of these women. Among women with HF, the use of potentially embryo-fetal toxic HF medications was low and decreased during pregnancy, in accordance with recommendations in drug labeling. Based on the low use of HF medications generally, and the population for which vericiguat is indicated (subtype of HF patients [symptomatic chronic heart failure with reduced ejection), vericiguat exposure is expected to be low. This information contributed to the review team’s determination that labeling would provide sufficient information to ensure the benefits of vericiguat outweigh its risks.
Evidence suggested that COVID-19 infection may induce a hypercoagulable state resulting in arterial thromboembolism (ATE) or venous thromboembolism (VTE). However, studies examining thrombotic complications from COVID-19 to date had included small samples, rarely included a comparator group, and had not evaluated characteristics associated with these outcomes. FDA initiated this study in the Sentinel System to determine the 90-day incidence of hospitalized ATE and VTE in patients with COVID-19 and subsequent 30-day mortality, identify risk factors for ATE and VTE events, and compare the incidence of ATE and VTE among patients with COVID-19 vs. patients with influenza. Patients were evaluated separately based on the setting (outpatient vs. inpatient) and timing (before vs. during COVID-19 vaccination availability) of their COVID-19 diagnosis.
Among patients hospitalized with COVID-19, the 90-day absolute risks of ATE and VTE were 15.8%-16.3% and 9.5%-10.9%, respectively (range represents periods before and during vaccine availability). Compared with patients with influenza, the risk of ATE was not significantly higher among patients with COVID-19 either before or during vaccine availability; however, risk of VTE was significantly higher among patients with COVID-19 before (HR 1.60; 95% CI: 1.43-1.79) and during vaccine availability (HR 1.89; 95% CI: 1.68-2.12). Among those with an ATE or VTE event, 30-day mortality was significantly higher in patients with inpatient-diagnosed COVID-19 versus influenza both before vaccine availability [(ATE: HR 3.45; 95% CI: 2.68-4.45) (VTE: HR 2.96; 95% CI: 1.84-4.76)] and during vaccine availability [(ATE: HR 3.45; 95% CI: 2.69-4.44) (VTE: HR 3.80; 95% CI: 2.41-6.00)].
Among patients with ambulatory-diagnosed COVID-19, the 90-day absolute risks of hospitalized ATE and VTE were 1.01%-1.06% and 0.73%-0.88%, respectively. Compared with patients with influenza, the risk of ATE was higher among patients with COVID-19 both before (HR 1.53; 95% CI: 1.38-1.69) and during vaccine availability (HR 1.69; 95% CI: 1.53-1.86). Risk of VTE also was higher among patients with COVID-19 before (HR 2.86; 95% CI: 2.46-3.32) and during vaccine availability (HR 3.56; 95% CI: 3.08-4.12). Among those with an ATE or VTE event, 30-day mortality was significantly higher in patients with ambulatory-diagnosed COVID-19 versus influenza both before vaccine availability [(ATE: HR 2.65; 95% CI: 1.88-3.73) (VTE: HR 2.36; 95% CI: 1.34-4.18)] and during vaccine availability [(ATE: HR 2.53; 95% CI: 1.82-3.51) (VTE: HR 2.58; 95% CI: 1.48-4.50)]. The results from this study informed FDA’s public health response during the COVID-19 pandemic.
On April 5, 2022, the Biden Administration issued a Presidential Memorandum directing the Secretary of Health and Human Services (HHS) to coordinate a new effort across the federal government to develop and issue the first-ever interagency national research action plan on “long COVID,” a term used to describe the long-lasting effects of having been infected with COVID-19. In response, FDA’s Office of Surveillance and Epidemiology (OSE) formed a working group tasked with outlining OSE's research plan on long COVID for fiscal years 2023-2024. As part of these efforts, an analysis was conducted to explore the use of FDA Sentinel’s electronic health record (EHR) data sources (specifically TriNetX) to identify and characterize long COVID, specific outcomes associated with long COVID, and relevant covariates (e.g., COVID-19 vaccination, prior COVID-19 infection, and treatments received during the acute disease phase). Results found under capture of long COVID cases in EHR data, with the most frequent signs/symptoms being in line with what has been reported in the literature.
Generic mixed amphetamine salt (MAS) products, particularly the immediate-release tablet products of specific generic MAS products, have been the subject of spontaneous reports to the Drug Quality Report System, a subset of the Food and Drug Administration's (FDA) MedWatch. Most complaints described lack of effectiveness and a need for a dose increase after switching to specific generic MAS products from other MAS products. The objective of these Sentinel analyses was to examine utilization and switching patterns among adults with a diagnosis of attention-deficit/hyperactivity disorder (ADHD) and/or narcolepsy, treated with immediate release MAS formulations in postmarketing real world settings. Results from this descriptive analysis suggest that switching appeared to correlate with the number of dispensings and the patterns of switching did not suggest increased switching away or switching back after use of the generics of interest. Based on these data, FDA determined that no regulatory action is needed at this time.
The FDA initiated this Sentinel study to determine if modafinil/armodafinil exposure in utero was associated with an increased risk of major congenital malformations. Some, but not all previous studies have reported an association between in utero exposure to modafinil/armodafinil and increased risk of major congenital malformations. Data from the Nuvigil (armodafinil)/Provigil (modafinil) pregnancy registry suggested potential safety signals of cardiac defects, overall congenital malformations, and musculoskeletal defects.
This study found that modafinil/armodafinil exposure in utero was not associated with cardiac malformations. Additionally, there was no association between modafinil/armodafinil exposure in utero and the risk of non-cardiac, musculoskeletal, or urinary malformations.
The FDA is considering whether the results of this study require any regulatory action.
Drug-drug interactions are an important clinical concern. In a March 2017 Advisory Committee, briefing documents raised the possibility that oxymorphone may be used for a particular niche - patients taking multiple medications - because its metabolism did not involve the hepatic cytochrome P450 system. FDA conducted an analysis to assess whether oxymorphone’s metabolism influenced prescriber practices. Results revealed that similar proportions of patients were dispensed oxymorphone combined with other CYP modifiers relative to reference products, suggesting that drug metabolism differences did not influence prescribing behavior. These results contributed to regulatory discussions about the use of oxymorphone drugs, including reformulated Opana ER (oxymorphone hydrochloride) that was withdrawn from the market for public health safety reasons.
The U.S. Food and Drug Administration (FDA) initiated this study in the Sentinel System to assess for immunosuppressive medications, which, as a side effect, reduce the body’s tumor surveillance and increase the risk of developing a malignancy. Over the last several years, the FDA has approved multiple immunosuppressive therapies for psoriasis and multiple sclerosis (MS). In this study, FDA explored the Sentinel System’s ability to surveil for malignancy by focusing on describing the duration of follow-up time after incident use of immunosuppressive medications used to treat psoriasis and MS, as well as the duration of therapy. This study also described the duration of follow-up for patients with prevalent Crohn’s disease, MS, psoriasis, and ulcerative colitis diagnoses.
Results indicated that for most immunosuppressive medications, greater than 60% of patients had follow-up time less than three years; less than 1% had greater than ten years of follow-up time. These results were further stratified by age groups and calendar year. Among patients with psoriasis, MS, or inflammatory bowel disease (IBD), the majority (greater than 65%) had less than three years of follow-up time.
Given that this study found a minority of patients had follow-up time of more than three years, the Sentinel System may have limited ability to detect malignancies that take several years to develop. This study helped understand the capabilities regarding conducting analyses using administrative claims for long-latency outcomes including malignancies.
The U.S. Food and Drug Administration (FDA) initiated this methods development study to test a new Sentinel System analytic tool to perform Interrupted Time Series (ITS) analyses for assessing the impact of FDA regulatory actions on drug utilization. The newly developed analytic tool and the impact of FDA's February 2010 and June 2010 Drug Safety Communications (DSCs) concerning the safety of using long-acting beta-2 agonists (LABA) alone without the use of long-term asthma controller medications in adult asthma patients were assessed. Findings from this test case demonstrated the successful development of the ITS tool. Study results showed that the initiation of LABA alone declined among asthma patients aged 18-45 years before the FDA DSC in June 2010 (-0.10% per quarter; 95% CI: -0.11% to -0.09%), and the downward trend continued after the FDA DSC. These findings were consistent with FDA’s previous LABA studies published in 2016 and 2017.
The reusable analytic tool can be applied to real-world databases formatted to the Sentinel Common Data Model for assessing the impact of FDA regulatory actions on drug utilization.
The FDA initiated this analysis to better understand utilization of hydroxyprogesterone caproate (HPC) injection, including Makena® and its generics, among pregnant women in the United States. We identified 3,445,739 live-birth pregnancies (among 2.9 million women) between 2008 and 2018 in the Sentinel Distributed Database (SDD). Of these pregnancies, 6.5 per 1,000 pregnancies used injectable HPC, showing modest use of injectable HPC during the second and/or third trimesters among all live-birth pregnancies in the SDD. The majority (73%) of pregnancies with injectable HPC use had at least one of three obstetrical indications of interest recorded before or during the pregnancy.
Sentinel analysis results were presented at an FDA Advisory Committee for consideration in assessing the potential public health impact of withdrawing Makena’s accelerated drug approval. Following the FDA Advisory Committee, the FDA’s Center for Drug Evaluation and Research (CDER) recommended withdrawing Makena’s accelerated approval. FDA withdrew approval of Makena on April 6, 2023, because the confirmatory trial failed to verify the clinical benefit of Makena nor did the trial show that Makena reduced the risk of preterm birth.
Recent estimates showed that patients aged 18 years or younger comprised approximately 14% of the population in the Sentinel Distributed Database (SDD). To conduct studies among pediatric patients that could support regulatory decisions, it is vital to understand the composition of this population. The FDA initiated this study to better understand the characteristics of this population, including demographics, vaccination status, medical comorbidities, major congenital malformations, medication administration, prescription claims, healthcare utilization, and available follow-up time in the SDD from January 2000 to May 2023.
Results indicate that the total volume of pediatric enrollees in the SDD from 2000-2023 ranged from about 16 million newborns (0-28 days) to about 46 million older children (6-11 years of age), with representation from all 50 U.S. states’ commercial and Medicaid health plans. The data span more than 20 years, and more than half of pediatric patients are continuously enrolled in their health plan for at least one year. The SDD is a valuable data source that can support investigations of pediatric medication exposures and health outcomes.
Since 1970, cannabis within the federal Controlled Substances Act’s (CSA’s) definition of “marijuana” or “marihuana” has been controlled under Schedule I of the CSA. Schedule I drugs have no currently accepted medical use in treatment in the United States. Despite control at the federal level, an increasing number of state, local, tribal, and territorial bodies have enacted laws allowing medical or both medical and nonmedical adult use of what is federally considered "marijuana" by the CSA’s definition. The Center for Drug Evaluation and Research (CDER) pursued analyses in the Sentinel System to assess the temporal trends in cannabis-related healthcare encounters, including cannabis-related disorders and poisonings, among commercially insured adults aged 18-64 years. Results showed that between 2017-2022, increasing trends in cannabis-related healthcare encounters were observed, largely in emergency departments and outpatient care settings. Of note, given the administrative nature of the data, reason for use or distinction between marijuana, synthetic cannabinoids, and federally legal hemp could not be determined. Sentinel results were cited in the 2023 scientific and medical evaluation of “marijuana” conducted by FDA on behalf of the Department of Health and Human Services (HHS) and transmitted to the Drug Enforcement Administration (DEA). The HHS evaluation provided a basis for the DEA’s proposed rule, published in the Federal Register (89 FR 44597, May 21, 2024), to reschedule “marijuana” from Schedule I to Schedule III of the CSA.
The FDA initiated this study in Sentinel to explore the use of TreeScanTM to support safety signal identification for baloxavir. Baloxavir is a first-in-class influenza-specific endonuclease inhibitor approved for acute uncomplicated influenza or post-exposure prophylaxis in adult and pediatric patients. TreeScan is a signal identification approach that scans thousands of health outcomes simultaneously while adjusting for multiple scenarios that can be used to monitor the underlying assumption of no clinical differences.
The goal of the study was to understand if there is an increased risk of safety outcomes in patients who received baloxavir versus in patients who received the comparator drug oseltamivir, a first-line influenza treatment and prophylactic having a different mechanism of action and greater postmarketing exposure than baloxavir. This study did not identify new safety issues with baloxavir as treatment or prophylaxis for influenza compared to oseltamavir in a large sample of real-world adult and pediatric patients, adding to the larger body of safety evidence for baloxavir.
The increasing use of genomic tests for diseases has great potential to improve health outcomes because of the trend toward targeted therapeutics such as personalizing treatments by genotype. Prescribing information for FDA-approved drugs may contain information on genomic biomarkers that can identify responders and non-responders to medications. However, few assessments on the use of genomic tests have been completed, because of the limitation of the existing administrative healthcare data, given the laboratories have traditionally billed for genetic testing using stacking codes that describe each step of the procedure required to perform a genetic test. Therefore, the goal of this study was to assess the feasibility of identifying genetic tests, their rates of use, and baseline characteristics of tested patients in the Sentinel Distributed Database (SDD).
Using the SDD for the time period of January 1, 2013, to December 31, 2015, FDA estimated the number of incident users of cetuximab, panitumumab, trametinib, dabrafenib, vemurafenib, cobimetinib, afatinib, erlotinib, tagrisso, gefitinib, dasatinib, imatinib, bosutinib, nilotinib, and ponatinib, and the utilization of five different genetic tests of the respective genes V-Ki-ras2 Kirsten rat sarcoma viral oncogene (KRAS), v-raf murine sarcoma viral oncogene homolog B1 (BRAF), epidermal growth factor receptor (EGFR), breakpoint cluster region-abelson (BCR-ABL), and the breast cancer susceptibility gene (BRCA) were estimated. Cetuximab and panitumumab were reported in 5,220 new users (0.08 new users/1000 eligible members). Dasatinib, imatinib, bosutinib, nilotinib, and ponatinib were reported in 3,545 new users (0.06 new users/1000 eligible members). The KRAS, BRAF, EGFR, BCR-AB and BRCA genetic tests were received by 5,210, 4,907, 4,730, 5,360, and 23,111 participants respectively. These results informed the FDA about the feasibility and limitations of using the Sentinel System and existing tools to assess the use of genetic testing.
The FDA initiated this study in the Sentinel System to gather data on drug utilization of angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs) in various subgroups of patients aged less than one year. Product labeling for renin-angiotensin system (RAS) inhibitors, including ACE inhibitors and ARBs, has varying recommendations for indicated pediatric populations (i.e., enalapril is approved for patients aged greater than one month; candesartan is approved for patients aged greater than one year, etc.). RAS inhibition causes fetal and neonatal abnormalities when used late in pregnancy by the mother and the effects of inhibition on the developing human kidney (during the first year of life) are unknown. The FDA sought to quantify the degree of prescribing of products affecting the RAS system in various pediatric age groups. Results of this study estimated the number of prevalent users and dispensings of ACE inhibitors and ARBs among these pediatric populations. This study provided pediatric drug utilization data that helped inform the review of some applications for ACE inhibitors and ARBs.
The purpose of this study was to assess whether Active Risk Identification and Analysis (ARIA) system tools could replicate the results of prior protocol-based assessments (PBAs) that evaluated acute myocardial infarction (AMI) and hospitalized heart failure risk following saxagliptin or sitagliptin use in comparison to long-acting insulins, pioglitazone, and second-generation sulfonylureas in the Sentinel Distributed Database (SDD). The study found that Sentinel modular programs were able to reproduce findings of prior PBAs which did not indicate an increased risk of AMI or hospitalized heart failure associated with saxagliptin or sitagliptin exposure.
Wixela Inhub (fluticasone propionate/salmeterol xinafoate) was the first approval for a generic inhaled product referencing Advair Diskus and was a significant milestone in establishing a bioequivalence pathway for future generic inhaled pulmonary products. FDA sought to describe the use of Advair Diskus and other brand inhaled corticosteroid (ICS) and long-acting beta-agonist (LABA) combination products in patients with chronic obstructive pulmonary disease (COPD) or asthma and switching patterns between Advair and other ICS/LABA products, including Wixela Inhub and the authorized generic (AG) for Advair Diskus.
Results showed substantial use of multiple branded ICS/LABA combination products in Sentinel, with Advair Diskus and Symbicort being the most common in asthma and COPD patients. There were minimal differences in the characteristics across users of different ICS/LABA products. Only a small proportion of Advair Diskus users switched to another ICS/LABA product during the study period. Switching was more common in prevalent users than new users. Switch backs were rare, with the most common patterns being patients switching from Advair Diskus to Wixela Inhub or Advair AG and back to Advair Diskus.
Based on the available findings, FDA determined that no regulatory action was needed.
Postmarketing surveillance of medical products entails an assessment of both the safety of treatments among cancer patients and the risk of malignancies associated with non-cancer treatments. To better inform feasibility for future studies to address these questions, FDA initiated this study to describe the duration of follow-up among patients with new-onset cancer diagnoses in Sentinel. Sample sizes and patterns of enrollment surrounding new-onset diagnoses of nineteen cancers were analyzed. Prior to a cancer diagnosis, the median enrollment ranged from 2.3-3.6 years (among those with a required one-year minimum enrollment), depending on cancer type. Following a cancer diagnosis, the median enrollment ranged from 0.5-2.3 years. Enrollment time surrounding cancer diagnosis may provide sufficient duration of follow-up for studies in which 1) cancer is the outcome, for drug-related adverse events of relatively short latency, or 2) cancer diagnosis is a component of the cohort eligibility.
International Classification of Diseases, Tenth Revision, Clinical Modification (ICD-10-CM) diagnosis codes are thought to have better accuracy for capturing suicide intent compared to International Classification of Diseases, Ninth Revision, Clinical Modification (ICD-9-CM) codes. As an update to a previous Sentinel study that used ICD-9-CM codes to identify intentional self-harm, this study assessed the risk of intentional self-harm events in patients aged 10 and older with asthma following use of either montelukast or inhaled corticosteroids (ICS) as monotherapy. Intentional self-harm in this study was defined using ICD-10-CM diagnosis codes, and the analysis used inverse probability of treatment weighting to balance baseline covariates. Among 752,230 and 724,855 patients in the montelukast and ICS exposure groups respectively, no association was found between montelukast use and intentional self-harm compared to ICS use [Hazard Ratio (95% Confidence Interval): 0.96 (0.85, 1.08)]. This finding was consistent across age, sex, psychiatric history and pre/post Boxed Warning period subgroups, and sensitivity analyses showed similar null findings. These results cannot exclude other neuropsychiatric idiosyncratic reactions to montelukast. Compared to the previous Sentinel study, this study identified about double the rate of intentional self-harm events, suggesting a greater sensitivity of ICD-10-CM codes for measuring intentional self-harm than ICD-9-CM codes. This study provided more information to FDA on the use of diagnosis codes for capturing intentional self-harm, an important health outcome in drug safety studies.
FDA initiated this feasibility study in the Sentinel System to explore the performance of TreeScan™ using International Classification of Diseases, Tenth Revision, Clinical Modification (ICD-10-CM) data. TreeScan™ is a signal identification approach that scans thousands of health outcomes simultaneously while adjusting for multiple scenarios that can be used to monitor the underlying assumption of no clinical differences.
The alert patterns observed in this feasibility study were highly similar to a prior analysis that evaluated Angiotensin-Converting Enzyme (ACE) Inhibitors and Angiotensin II Receptor Blockers (ARBs) using International Classification of Diseases, Ninth Revision, Clinical Modification (ICD-9-CM) as the Tree and propensity score covariates. Based on these results, FDA determined the ICD-10-CM Tree and propensity score covariates are ready to use for signal identification (i.e., TreeScan analyses).
This study was previously conducted in the Mini-Sentinel pilot program to assess the benefit-risk profile of using Eliquis (apixaban) compared to warfarin in patients with atrial fibrillation (AF). The original analysis suggested a signal of harm for apixaban use related to the health outcome of intracranial hemorrhage (ICH). However, the number of apixaban users in the original analysis was low as apixaban was approved in 2014. Therefore, an updated study with increased sample size was required. This study found that in AF patients of all ages initiating either apixaban or warfarin for stroke prevention, apixaban was associated with a decreased risk of gastrointestinal (GI) bleeding, ICH, and ischemic stroke compared with warfarin. No regulatory action was required as findings suggested a favorable benefit-risk profile for apixaban compared to warfarin for stroke prevention in patients with AF.
On July 7, 2015, sacubitril/valsartan (SV) was approved for the treatment of heart failure in patients with systolic dysfunction. Excess risk of angioedema, a rapid-onset, transient, potentially life-threatening swelling of the tongue, lips, mouth, throat, nose, and other parts of the face, has been described among users of angiotensin-converting enzyme inhibitors (ACEI), angiotensin receptor/neprilysin inhibitors (ARNI) and angiotensin receptor blockers (ARB). Combining ACEIs and ARNIs has been shown to be associated with a greater risk of angioedema compared to ACEIs use alone.
Therefore, due to the potential higher risk of angioedema when combining ARNI (sacubitril) and ARB (valsartan), compared to the use of sacubitril or valsartan alone, FDA compared the incidence of angioedema (serious and non-serious events) among the users of SV, and other drug classes indicated for heart failure, such as ACEIs, ARBs, β-blockers and digoxin in the Sentinel Distributed Database (SDD).
No increased risk of angioedema was identified among SV new users compared with ACEI or ARB users. However, there was an increased risk of angioedema among SV users who had recently switched from an ACEI or ARB compared with SV new users. As the labeling was consistent with the study results, FDA determined no additional regulatory action is needed at this time.
Sufficient observable person-time in the indicated population is necessary for conducting drug safety analyses. While the median length of observation time for members in commercial insurance claims databases is typically less than two years, it is unknown if individuals with some chronic conditions have different lengths of observation time in these data sources. The objective of this study was to assess prevalence and duration of follow-up for 24 chronic condition cohorts in the Sentinel Distributed Database (SDD), as defined using the Centers for Medicare and Medicaid Services’ (CMS) Chronic Conditions Data Warehouse condition algorithms. Hypertension and hyperlipidemia had the highest prevalence (26.3% and 23.9%), while all five cancers assessed had low prevalence (0.2 to 1.5%). Median follow-up ranged from 0.8 (lung cancer) to 2.7 years (hyperlipidemia). As this query intended to characterize the data in the SDD, no regulatory actions were taken.
Alosetron (brand name Lotronex) is approved for the treatment of women with severe diarrhea-predominant irritable bowel syndrome (IBS-D). U.S. Prescribing Information for alosetron presents a boxed warning for serious gastrointestinal adverse reactions, including ischemic colitis. In addition, the Food and Drug Administration (FDA) required a Risk Evaluation and Mitigation Strategy (REMS) program for Lotronex and approved generics to mitigate the risks of ischemic colitis and serious complications of constipation.
To support an evaluation of the REMS programs for Lotronex and alosetron, FDA conducted an analysis in the Sentinel System to provide contemporary estimates of the incidence of ischemic colitis during alosetron use. Among women without pre-index intestinal ischemia, the incidence of ischemic colitis was estimated to be 3.08 (95% confidence interval, 1.80-5.26) per 1,000 in the first six months after a new pharmacy dispensing for alosetron. The estimated rate of ischemic colitis was consistent with that described in the alosetron Prescribing Information. These data supported FDA’s decision to eliminate the Lotronex and alosetron REMS programs.
In 2017, the FDA approved Dupixent (dupilumab), an interleukin-4 receptor alpha agonist. Dupixent is currently indicated for atopic dermatitis for patients aged six months and older, asthma for patients aged six years and older, chronic rhinosinusitis with nasal polyposis in adult patients, eosinophilic esophagitis in patients one year old and older, and prurigo nodularis in adult patients. FDA sought to understand whether Dupixent was being used off-label prior to the eosinophilic esophagitis approval, and if so, to what extent.
An analysis was completed in Sentinel in 2021 to investigate utilization patterns of Dupixent by varying cohort inclusion requirements with broad and narrow definitions of eosinophilic esophagitis. Overall, findings suggested that very few patients used Dupixent to treat eosinophilic esophagitis. As this analysis was conducted for informational purposes only, FDA did not take regulatory action.
A U.S. Food and Drug Administration (FDA) Advisory Committee (AC) meeting was planned to discuss the application for a new Penicillin Allergy Skin Test Kit (Pre-Pen Plus) product in March 2019. In preparation for the AC meeting, FDA used Sentinel to consider background information on trends in the recent use of penicillin allergy skin testing generally. The Sentinel analysis provided an estimate of the increasing trend in Pre-Pen penicillin allergy testing between 2008 and 2018. Ultimately, for reasons unrelated to the Sentinel analysis, FDA canceled the planned AC meeting.
Following the new regulatory pathway for approval of biosimilar and interchangeable products, FDA needs to be able to evaluate their post-marketing experience. However, how biosimilars are coded for reimbursement, and therefore appear in claims data, is unknown. Since accurate identification of biologics and biosimilars in claims data is a fundamental need for future studies of these products, this study was conducted to examine how patient exposure to biologics and biosimilars can be identified in claims data. Use of filgrastim and infliximab was identified primarily via Healthcare Common Procedure Coding System (HCPCS) codes (filgrastim: 86.4%-97.7%; infliximab: 87.8%-100%) although some use was identified via National Drug Codes (NDCs) (filgrastim: 2.2%-13.5%; infliximab: <0.1%-6.5%). The median exposure episode gap ranged from 1 to 3 days for filgrastim and 48 to 50 days for infliximab. Data on identification of biosimilars in claims data and observed gaps between exposure episodes can be used to support future drug safety studies of biosimilars.
Percutaneous transluminal septal myocardial ablation (PTSMA) is an alternative to surgery for left ventricular outflow tract obstruction in patients with hypertrophic obstructive cardiomyopathy. PTSMA involves injection of dehydrated alcohol into the coronary artery, inducing a localized basal septal myocardial infarction which remodels the outflow tract. Ablysinol, a dehydrated alcohol product, was approved by FDA on June 21, 2018. FDA wanted to understand how approval of this new dehydrated alcohol product would impact the number of PTSMA procedures conducted and subsequent post-procedural complications.
FDA conducted a study in the Sentinel System to evaluate changes in the frequency of PTSMA procedures conducted and changes in the occurrence of potential post-procedural complications after approval. After Ablysinol approval in 2018, the frequency of PTSMA procedures per year, the proportion of potential post-procedural complications such as heart failure, atrioventricular block, and permanent pacemaker placement observed, and the proportion of PTSMA procedures requiring a repeat PTSMA procedure, increased only slightly in this study. Evaluation of death was not pursued in this analysis due to under capture in the assessed data. The FDA determined that no regulatory action is needed at this time.
FDA initiated this pilot study in Sentinel to explore the use of TreeScanTMfor routine drug safety surveillance, using a use case of Ozempic (a specific form of semaglutide). TreeScan is a signal identification approach that scans thousands of health outcomes simultaneously while adjusting for multiple scenarios that can be used to monitor the underlying assumption of no clinical differences.
The study specifically sought to monitor non-pregnancy and non-cancer outcomes among new users of Ozempic compared to new users of sitagliptin among patients with type 2 diabetes. FDA determined the statistical alerts generated by the TreeScan analysis do not warrant further evaluation or action. This pilot demonstrated that tree-based scan statistics can be applied to the Sentinel Distributed Database using an active comparator design to simultaneously evaluate thousands of safety outcomes for medicines as an additional safety surveillance tool.
FDA initiated this pilot study in Sentinel to explore the use of TreeScanTM for routine drug safety surveillance, using a use case of Aimovig (erenumab). TreeScan is a signal identification approach that scans thousands of health outcomes simultaneously while adjusting for multiple scenarios that can be used to monitor the underlying assumption of no clinical differences.
The study specifically sought to monitor non-pregnancy and non-cancer outcomes among new users of Aimovig using a self-controlled risk interval analysis and tree-based scan statistics. FDA determined the statistical alerts generated by the TreeScan analysis do not warrant further evaluation or action. This pilot demonstrated that tree-based scan statistics can be applied to the Sentinel Distributed Database (SDD) using a self-controlled risk interval design to simultaneously evaluate thousands of safety outcomes for medicines as an additional safety surveillance tool.
In June 2020 the United Kingdom Randomized Evaluation of COVID-19 Therapy (RECOVERY) trial, reported benefit from dexamethasone use in severely ill hospitalized patients with COVID-19, but potential harm in patients not requiring oxygen supplementation. As a result, in October 2020, the National Institutes of Health (NIH) issued guidelines advising against the use of systemic corticosteroids in non-hospitalized COVID-19 patients. The Rapid Sentinel Distributed Database (SDD) was used to describe the demographic and clinical characteristics of U.S. non-hospitalized patients with COVID-19 who initiated systematic corticosteroids and those who did not from April 2020 to July 2021. Additionally, descriptive information was generated on the proportion of corticosteroid initiators who were hospitalized within 30 days of initiation.*
There were 766,105 eligible COVID-19 patients (median age 48.5 years [standard deviation, 19.9 years]), 9.4% of whom received corticosteroids in an outpatient setting within 14 days of COVID-19 diagnosis. Treatment often started on the day of COVID-19 diagnosis (51.3%), largely through pharmacy dispensings rather than during medical encounters. Corticosteroid use increased over time from 2.2% initiating in April 2020 to 13.8% initiating in July 2021. Among those who received corticosteroids in an outpatient setting, 11.1% were hospitalized (COVID-19 in any diagnostic position) within 30 days of initiation.
An early version of these analyses was presented to the NIH COVID-19 Treatment Guidelines Panel (April 2021). Later, updated analyses were published in the Journal of the American Medical Association (JAMA) (April 2022). The Centers for Disease Control and Prevention (CDC) subsequently issued a health advisory citing these data and alerting physicians and the public that systemic corticosteroids are not recommended for patients with mild to moderate COVID-19 who do not require supplemental oxygen.
*In addition to the Rapid SDD, the FDA conducted parallel analyses in three other data sources. Results here pertain only to Sentinel.
In May 2013, the FDA approved golimumab for ulcerative colitis with a post-market required (PMR) study to assess risk of lymphoma. In response to concerns about low enrollment in this study, the FDA conducted an analysis in Sentinel's Merative™ MarketScan® Research Databases using the Query Builder tool to understand if golimumab utilization in patients with ulcerative colitis (UC) was contributing to low patient enrollment. From January 2017 – December 2019, the relative frequency of anti-TNFα new-use episodes for UC in adults was 56%, 39%, and 5% for adalimumab, infliximab (including biosimilars), and golimumab, respectively. These data confirmed low utilization of golimumab and supported the FDA’s decision to discontinue further enrollment in the PMR study.
The Bipartisan Budget Act of 2018 (P.L. 115-123) included a provision for the Government Accountability Office (GAO) to review the prevalence of obesity and the use of obesity drugs. GAO requested that FDA assess utilization patterns and treatment duration for the nine available prescription used to treat obesity. FDA used the Sentinel System to assess the baseline characteristics of patients initiating weight management drugs and evaluate the duration of treatment of the first treatment episode and cumulative duration across all treatment episodes. These results were incorporated into the GAO report entitled, “Few Adults Used Prescription Drugs for Weight Loss and Insurance Coverage Varied.”
FDA initiated this pilot study in Sentinel to explore the use of TreeScanTM for routine drug safety surveillance of biosimilars, using a use case of Zarxio (Filgrastim-sndz). Biosimilars are biologic medications that are highly similar to existing approved biologics with no identified clinically meaningful differences in safety, effectiveness, and quality. However, the complex nature of biologics generally means a biosimilar is not identical to the reference product. TreeScan is a signal identification approach that scans thousands of health outcomes simultaneously while adjusting for multiple scenarios that can be used to monitor the underlying assumption of no clinical differences.
The study specifically sought to monitor non-pregnancy and non-cancer outcomes among new users of Zarxio compared to new users of Neupogen. FDA determined the statistical alerts generated by the TreeScan analysis do not warrant further evaluation or action. This pilot demonstrated that tree-based scan statistics can be applied to the Sentinel Distributed Database to simultaneously evaluate thousands of safety outcomes for biosimilar medicines as an additional surveillance tool.
Epidiolex (prescription cannabidiol) was approved in June 2018 for the treatment of seizures associated with Lennox Gastaut syndrome (LGS), Dravet syndrome (DS), or tuberous sclerosis complex (TSC) in patients 1 year of age and older.
As a part of FDA’s mission to perform surveillance of cannabidiol-containing products, a utilization analysis was conducted in Sentinel’s Merative™ MarketScan® Research Databases to inform feasibility of future studies.
Based on the available findings, FDA determined that no regulatory action was needed.
In March 2020, insulins were transitioned from being regulated as drugs to being regulated as biological products. This transition allows for the introduction of biosimilars, which are highly similar to, and have no clinically meaningful differences from, their reference biological medication. To facilitate pharmacovigilance, an analysis was conducted to characterize how insulin was captured in electronic healthcare data sources, specifically via National Drug Codes (NDC) or Healthcare Common Procedure Coding System (HCPCS) codes. This analysis demonstrated that over 98% of the 107 million insulin claims in Sentinel between 2013 and 2018 were identified using NDC codes, supporting the use of NDCs to identify insulin exposure in drug safety studies using electronic healthcare data. Having a variety of different tools to identify products for pharmacovigilance is of high value. We have demonstrated that a large fraction of insulin use in Sentinel can be identified using NDCs. That, in addition to other identifiers, supports effective pharmacovigilance.
In response to a sponsor’s reported difficulty in accruing patients for a postmarket required study (PMR) using administrative healthcare data to retrospectively assess pregnancy outcomes in a cohort of women exposed to ixekizumab, the FDA conducted a Sentinel analysis to obtain contemporary information about the utilization of ixekizumab in pregnancy. Specifically, the study estimated the number of pregnancies with a pharmacy dispensing for ixekizumab and comparator drugs, overall and among patients with approved indications for ixekizumab. Between March 2016 and June 2021, there were 29 ixekizumab-exposed pregnancies, of which 19 had no concomitant exposure to comparators. For the 19 uniquely exposed pregnancies, use increased from no pregnancies in 2016 and 2017 to three pregnancies in 2018 (15.8%), two (10.5%) in 2019, and 10 pregnancies in 2020 (52.6%). Although the number of pregnancies exposed to ixekizumab is low, the analysis showed that the number of pregnancies exposed to ixekizumab is increasing over time. This contributed to FDA’s decision to continue evaluating potential risks associated with ixekizumab use during pregnancy via the pregnancy safety PMR.
In response to case reports describing musculoskeletal adverse events, including decreases in bone density, in young adults who had been exposed to leuprolide acetate during childhood, the FDA further evaluated the risk of fracture with the use of gonadotropin-releasing hormone (GnRH) agonists indicated for the treatment of central precocious puberty (CPP), including leuprolide acetate. The Sentinel analysis estimated the risk of major fractures among individuals with CPP who initiated leuprolide acetate during childhood and compared them with leuprolide acetate-unexposed age- and sex-matched reference populations with and without CPP. Compared separately to the leuprolide-unexposed children with or without CPP, the study observed a lower risk of fracture in female leuprolide users with CPP, but no statistically significant difference in fracture risk for male leuprolide users with CPP. Results from the post hoc analysis were consistent with the findings from the main analysis. Because results from this study provided no evidence for an increased risk of fracture following leuprolide use during childhood, FDA determined that no regulatory action is needed at this time.
Gadolinium-based contrast agents (GBCA) are used in magnetic resonance imaging (MRI) or magnetic resonance angiography (MRA) diagnostic imaging procedures to aid in the visualization of abnormalities. GBCAs have a warning for hypersensitivity reactions, and seizure risk is listed as a postmarketing adverse event. The FDA identified cases of seizures shortly following GBCA exposure in the FDA Adverse Event Reporting System (FAERS) case reports and initiated a Sentinel study to better understand the magnitude of seizure risk associated with exposure to GBCAs during MRI and MRA procedures. An increased risk of seizure was not observed comparing contrast to non-contrast MRI in this study (RR=1.04, 95% CI 0.62-1.61). FDA determined that current labeling was sufficient to communicate that seizures have been observed in the post-market setting.
Follow up investigation of case reports of cardiac events after long term stimulant use. FDA decided that no action is necessary at this time, based on available information.
Tardive dyskinesia (TD) is a known safety risk with use of metoclopramide. During the review of an application for a nasal spray formulation of metoclopramide for use in diabetic gastroparesis, FDA wanted to understand the duration of use, since the risk of developing TD increases with duration of treatment and cumulative dosage. To understand real-world use patterns of metoclopramide, FDA conducted an analysis on the treatment episode duration, number, frequency, and cumulative dose among users of other available metoclopramide dosage forms, which had labeled recommended durations of use of 8 weeks or less. Among women ages ≥ 40 years identified in the Sentinel Distributed Database between January 1, 2009 and September 30, 2015, there were over 1.4 million users of metoclopramide. Of the 3.7% of these women with diabetic gastroparesis, approximately 25% had metoclopramide treatment episodes longer than 4 weeks. These data provided context for FDA’s assessment of the application by providing information about the potential for the target population to use a nasal metoclopramide product outside the parameters supported by the available safety data.
This Sentinel analysis was initiated to determine length of therapy for oral metoclopramide among pediatric patients aged 0-17 years. Metoclopramide includes a boxed warning for increased risk of tardive dyskinesia (TD) for use > 12 weeks and is not recommended in pediatric patients due to risk of central nervous system complications. Based on the prescription claims data from 2012-2015, an estimated 10% of pediatric patients aged 0-17 years received prescriptions for oral metoclopramide for > 12 weeks, potentially increasing their risk of developing TD. These data provided context for FDA’s assessment of proposed pediatric use by providing information about the potential utilization outside the parameters supported by the available data.
This analysis provided information on concomitant utilization of ticagrelor and moderate/high-intensity statins. FDA received postmarket reports of statin-induced myopathy/rhabdomyolysis in elderly patients taking atorvastatin or rosuvastatin with ticagrelor. Considering that use of high-intensity statins is advised in secondary prevention following a myocardial infarction, FDA initiated this analysis to understand utilization patterns for patients concurrently taking ticagrelor and a statin and provide context for postmarket case reports. Our Sentinel analysis found that out of all ticagrelor 90mg exposures in patients ≥75 years, the proportion of concomitant use with high-intensity statins increased from 57% in 2012 to 66% in 2016. Based upon these data and analyses from other sources, FDA decided that no regulatory action was needed at this time.
Cases of severe uterine bleeding associated with use of novel oral anticoagulants (ACs) have been reported in the FDA Adverse Event Reporting System (FAERS) and the medical literature. FDA conducted a Sentinel study to examine severe uterine bleeding events requiring medical intervention in women treated with oral ACs. Among 1,050,192 new users of oral ACs, the incidence rates of severe uterine bleeding with medical, transfusion, and surgical (e.g., hysterectomy, myomectomy) management were 0.6, 1.7, and 5.0 per 1000 person-years, respectively. These findings contributed to the following class-wide label change for oral ACs in Section 8.3, “The risk of clinically significant uterine bleeding, potentially requiring gynecological surgical interventions, identified with oral anticoagulants including [PRODUCT name] should be assessed in females of reproductive potential and those with abnormal uterine bleeding.”
FDA conducted a study to further investigate a post-market clinical trial that identified an elevated risk of cardiovascular events. Sentinel was used to describe the real-world utilization of urate lowering therapies to help inform the committee’s determination that a population exists for whom the benefit-risk is favorable.
This analysis characterized the frequency of IV iron utilization by week relative to live birth and stillbirth deliveries. Information from this analysis contributed to a class-wide labeling update for parenteral iron products to add new safety information to the Use in Specific Populations, Pregnancy section of the label. This update describes the risk of severe adverse reactions to pregnant women and their fetus.
Use of PDE5 inhibitors was assessed among reproductive age women, including among pregnant women, to investigate a concern arising from an international clinical trial. FDA decided that no action is necessary at this time, based on available information.
In response to observational studies showing an elevated risk of non-melanoma skin cancer associated with hydrochlorothiazide (HCTZ) use, FDA assessed the risk of non-melanoma skin cancer for patients treated with HCTZ-containing products compared to non-HCTZ angiotensin-converting-enzyme inhibitor (ACEI)-containing products in the Sentinel System. FDA found HCTZ is associated with an increased risk of non-melanoma skin cancer. In the Sentinel study, increased risk was predominantly for squamous cell carcinoma and in white patients taking large cumulative doses. These findings informed additions to the Adverse Reactions and Information for Patients sections of the label.
This analysis provided information on use patterns of prescription ranitidine in patients with Medicare and private health care insurance. These data, in addition to data from other sources, were used to provide context for CDER’s Nitrosamine Impurities Task Force to better understand the use patterns among U.S. individuals. These data also informed the feasibility of a Sentinel analysis to evaluate clinical outcomes associated with prescription ranitidine use.
FDA presented a Sentinel study at a 2019 Advisory Committee of the risk of mental health side effects with montelukast compared to inhaled corticosteroids (ICS). In this study, FDA did not identify an elevated risk of hospitalized and treated outpatient depressive disorders, self-harm, and suicides among patients aged 6 years and older with asthma using montelukast compared to ICS. FDA also found no evidence that the risk of mental health side effects was modified by the 2008 FDA Early Communication. However, after reviewing the available information and convening a panel of outside experts, FDA strengthened the existing warnings about serious behavior and mood-related changes with montelukast by requiring a Boxed Warning because the benefits of montelukast may not outweigh the risks in patients with mild symptoms who could be adequately treated with other medicines.
FDA held an Advisory Committee (AC) meeting to discuss the definition of opioid-sparing and opioid-replacement analgesics, with particular attention to acute pain in the post-surgical setting. A Sentinel analysis was conducted to understand the variation in opioid use based upon surgical type. Sentinel data indicated that opioid exposure could be reduced among select surgical populations, particularly those undergoing less invasive procedures. However, the analysis also suggested that longer initial prescription durations might be appropriate in some cases and maintaining flexibility in prescribing options is essential to account for variation in opioid analgesic needs by patient as well as procedure.
Because the use of Zydelig in certain treatment regimens led to unacceptable levels of toxicity (pneumonitis, hepatitis, colitis), FDA evaluated the use of Zydelig concomitantly with other antineoplastic agents, in association with the labeled indication as well as other indications. The Sentinel analysis showed low observed rates of Zydelig concomitantly used with therapies which the labeled prescribing information describes as not indicated or not recommended. Based upon these data and analyses from other sources, FDA decided that no regulatory action was needed at this time.
FDA held an Advisory Committee (AC) meeting to better understand the use of higher dose opioid analgesics in the outpatient setting to inform a discussion of potential risk management strategies due to increasing public concern that these products may be more harmful in cases of accidental exposure and overdose, and may be more sought out for misuse and abuse. A Sentinel analysis was conducted to understand the current clinical use of higher dosage strength opioid analgesic products for pain management. Sentinel data indicated that patients on higher dosage strength opioid analgesic products had a higher prevalence of comorbidities, mental health disorders or substance abuse compared to patients on lower dosage strength opioid analgesic products. Moreover, the data showed that use of higher dosage strength products comprised a small proportion of all opioid use and has declined in recent years.
Feasibility assessment that supported an ARIA sufficiency determination to replace a sponsor postmarketing requirement (PMR) safety study for canagliflozin and renal cell carcinoma.
Based on preliminary results from an observational study suggesting a higher risk of neural tube defects among offspring of pregnant women using dolutegravir (see the FDA Drug Safety Communication below), FDA conducted a descriptive analysis of the prevalence and timing of dolutegravir use among HIV-infected women of childbearing age and HIV-infected women who had pregnancies with live birth deliveries. Prevalence of dolutegravir use among HIV-infected women of childbearing age was substantial, but the absolute number of dolutegravir-exposed pregnancies was very low. Although there was insufficient product exposure in pregnant women to support a robust safety assessment and the Sentinel mother-infant linkage was not yet available, the Sentinel analysis provided broader context regarding dolutegravir use among HIV-infected women who are of childbearing age or who had pregnancies with live birth deliveries.
Combined with evidence from the Centers for Medicare & Medicaid Services, risk of seizure was determined to be driven primarily by underlying comorbidities. FDA decided that no action is necessary at this time, based on available information.
Feasibility assessment of ARIA to conduct a postmarket safety study. FDA decided that no action is necessary at this time, based on available information.
As part of FDA’s routine review of postmarketing studies, FDA conducted a drug utilization analysis of TNF alpha inhibitors in pregnant women in the Sentinel System. The study found that among pregnant women with a chronic inflammatory condition (ankylosing spondylitis, juvenile idiopathic arthritis, psoriatic arthritis, psoriasis, rheumatoid arthritis), there was a preference to use etanercept compared to other TNF alpha inhibitors. For most TNF alpha inhibitors, use during pregnancy decreased after the first trimester. This assessment was one source of evidence considered for the Pregnancy and Lactation Labeling Rule (PLLR) Conversion Safety Labeling Change for Enbrel (etanercept).
Sentinel data was used to support decisions around potential labeling changes for antipsychotics and stroke risk. FDA decided that no action is necessary at this time, based on available information.